

Review
doi: 10.1021/ac5040242.
Epub 2014 Nov 14.
Applications of hydrogen/deuterium exchange MS from 2012 to 2014
Affiliations
- PMID: 25398026
- PMCID: PMC4287169
- DOI: 10.1021/ac5040242
Item in Clipboard
Review
Applications of hydrogen/deuterium exchange MS from 2012 to 2014
Anal Chem.
.
No abstract available
Figures
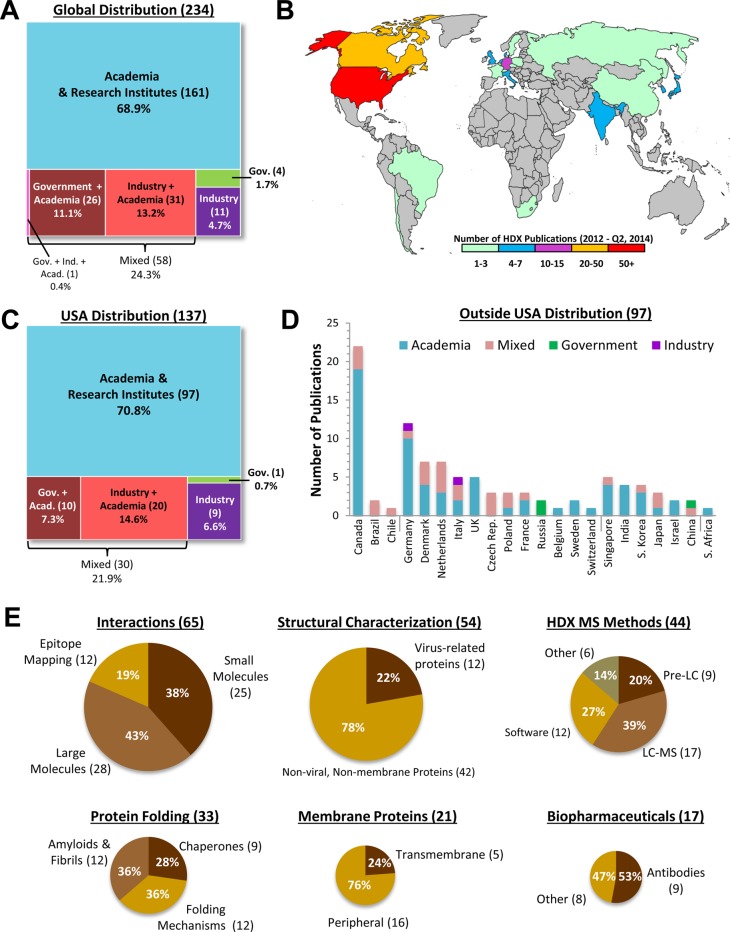
Synopsis of the published
applications of HDX MS from January 2012
to June 2014. (A) A total of two-hundred thirty-four (234) articles
were published and classified according to the scheme shown, based
on author affiliations. The number of articles in each category is
shown in parentheses; review articles were not included. (B) The global
distribution of the articles in panel A, based on the home institution
of the communicating author of each article. (C) Similar to panel
A, but for the largest source of articles, the United States of America.
(D) Breakdown of the non-USA publications according to country and
to sector. The data are divided roughly by continent going from left
to right. (E) The six topical classifications chosen for the Review
are shown as pie graphs, with the size of each pie equivalent to the
number of articles (shown in parentheses) in each topic. Further subdivision
within topics follows the order of the main text of the article.

Application to the study
of protein folding and unfolding. (A)
Example of determining the folding order of a protein with HDX MS,
here the order of α1-antitrypsin. Modified with permission from
ref (1). Copyright
2012 National Academy of Sciences of the United States of America.
(B) Example of monitoring protein unfolding via conformational dynamics.
Totally deuterated β2-microglobin (β2m) was incubated
in H2O buffer and intact mass spectra acquired at the times
shown. Exchange through EX1+EX2 kinetics (blue peaks), representing
a very unfolded unprotected form, was observed from which a rate of
EX1 unfolding could be extracted (right graph). (C) The EX1 rate of
β2m unfolding was measured for a host of other conditions, including
mutation and solution additives. Panels B and C modified and used
with permission from ref (12). Copyright 2012 John Wiley and Sons.

HDX MS in the
study of chaperone-assisted protein folding. The
TIM-barrel protein DapA was unfolded with denaturant and, upon dilution
of the denaturant, allowed to fold and assemble into its native tetramer
spontaneously (A) or in the presence of GroEL/ES (B). After various
periods of refolding, pulse labeling, pepsin digestion, and mass analysis
were performed. The protection half-times (colored by the categories
shown: red, yellow, blue) for segments of the protein (left) were
greatly accelerated by the chaperones compared to the spontaneous
folding. The chaperones also changed the order of folding. The locations
of each protection category are shown at the right on the crystal
structure of the assembled tetramer. Reprinted with permission from
ref (31). Copyright
2014 Elsevier.

Observing structural changes during function.
It was proposed that
the LOV (light–oxygen–voltage) domain protein VIVID
(VVD) underwent a structural change upon illumination and HDX MS was
used to show that this was the case. (A) Continuous labeling of the
dark state (VVDD) and light state (VVDL) revealed unfolding with EX1
kinetics and very different time scales of unfolding for the two states.
The location of the unfolding was determined with analysis of pepsin
fragments (example spectra shown in panel A inset). (B) Comparison
of deuterium levels in peptides and (C) interpretation of the data
on the structure of the protein and its dimer indicated that all structural
elements of VVDD incorporate more deuterium in nearly all regions
except for the N-cap. See ref (32) for full details. Modified with permission from ref (32). Copyright 2013 Elsevier.

Application of HDX MS to the study of viruses and virus-related
proteins. (A) Analysis of the binding to peripentonal hexons of the
33-residue N-terminal fragment of the precursor VI protein (pVIn)
from human adenovirus. Exchange into purified hexons was compared
with and without pVIn and the affected regions were localized. Modified
with permission from ref (72). Copyright 2014 Elsevier. (B) Binding-induced changes in
HIV Env trimers were determined when complexed with two different
small molecule HIV entry inhibitors (NBD-556 or BMS-806) that block
the CD4 binding site. Changes in exchange were compared to those seen
when Env trimmers were bound to CD4 alone (not shown here). Modified
with permission from ref (81). Copyright 2014 Elsevier.
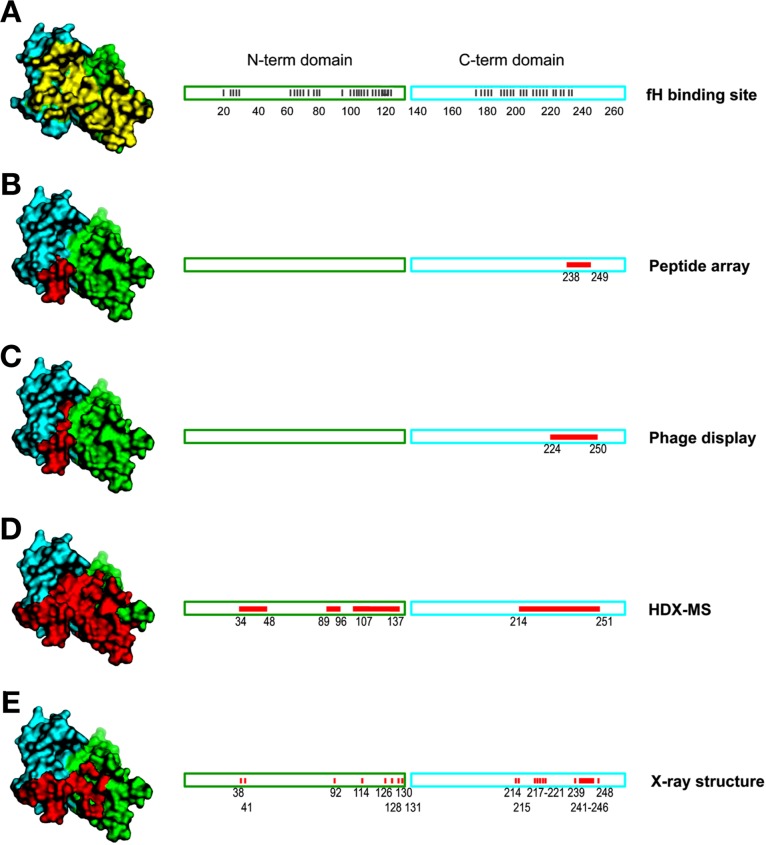
HDX MS for epitope mapping of a monoclonal antibody
(mAb) against
factor H binding protein (fHbp), a virulence factor, and vaccine antigen
of the causative agent of bacterial meningitis, Neisseria
meningitidis. Comparisons were made against (A) the known
interface between factor H (fH) and fHbp, interface residues colored
yellow in the structure at the left, and (B–E) epitope mapping
data, colored red on the structure and linear representation, for
mAb 12C1 and fHbp by various methods. HDX MS mapping (panel D) identified
all regions but not with the resolution of the cocrystal structure.
Reprinted with permission from ref (86). Copyright 2013 National Academy of Sciences
of the United States of America.

Example of mapping protein/protein interactions by HDX
MS. (A)
The effects of binding the nucleosome assembly protein 1 (Nap1) to
the histone H2A–H2B heterodimer were shown by comparing exchange
into the H2B portion of H2A–H2B alone (left) to exchange into
H2B when bound to Nap1 (middle). The regions most affected by binding
were mapped to the crystal structure of H2B (colored blue, right panel).
(B) Analysis by HDX MS suggested regions where mutations might be
made and tested in other assays; mutants 3 and 4 (residues changed
indicated in yellow and red, respectively) later showed reduced binding
to Nap1 in FRET assays. Modified with permission from ref (97). Copyright 2013 Elsevier.

Small molecule interactions with a protein. The AMP-activated
protein
kinase (AMPK) was incubated with various small molecules (A769662,
beta cyclodextrin, and staurosporine), and HDX MS was performed. Changes
in deuteration relative to unbound AMPK were both (A) mapped to the
crystal structure of AMPK and (B) displayed schematically. The percentages
of deuterium differences were mapped according to the key shown. Reprinted
with permission from ref (124). Copyright 2013 Elsevier.

Characterization of Fc fusion proteins and how their parts
relate
to naturally occurring versions. HDX MS of recombinant factor IX (rFIX)
was compared to a fusion of rFIX with an Fc of an antibody (termed
rFIX-Fc). The pattern of differences in deuterium levels of rFIX as
a result of calcium binding (A) was the same as that observed when
the fusion form, rFIX-Fc, bound to calcium (B) meaning that the FIX
portion was not impaired in its calcium binding activity by being
attached to the Fc. There were essentially no differences (C, D) between
exchange into rFIX and rFIX-Fc with calcium or without calcium. Reprinted
with permission from ref (158). Copyright 2012 Wiley.

Membrane
protein investigations with HDX MS. (A) Exchange into
phosphoinositide 3-kinase γ (also termed p110γ) was compared
to exchange during interactions with its regulatory/adaptor subunit
p101 (left). The complex of p110γ/p101 was then labeled with
and without empty liposomes (middle) or liposomes containing G-proteins
(Gβγ) (right) to ascertain the role of the membrane and
effects of binding. See ref (229) for full details. Reprinted with permission from ref (229). Copyright 2013 National
Academy of Sciences of the United States of America. (B) HDX MS of
BmrA, a bacterial multidrug ABC transporter, was performed while the
protein was in n-dodecyl-β-d -maltoside
(DDM) detergent. HDX results for analysis of an apo form were superimposed
on the 3D model of the open conformation (left), and analysis of a
closed form ATP-Mg-bound mutant were superimposed on a 3D model of
the closed conformation (right). Coloring is based on the percentage
of deuterium after 1 h of labeling, according to the color scale at
the right. Reprinted with permission from ref (181). Copyright 2012 National
Academy of Sciences of the United States of America.
References
-
- Jankowska E.; Stefanowicz P.; Sosnowska M.; Karpowicz P.; Radziszewska K.; Szewczuk Z.; Szymanska A. Proteins 2012, 80, 2417–2425. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
