

BRCA1 haploinsufficiency for replication stress suppression in primary cells
- PMID: 25400221
- PMCID: PMC4243249
- DOI: 10.1038/ncomms6496
BRCA1 haploinsufficiency for replication stress suppression in primary cells
Abstract
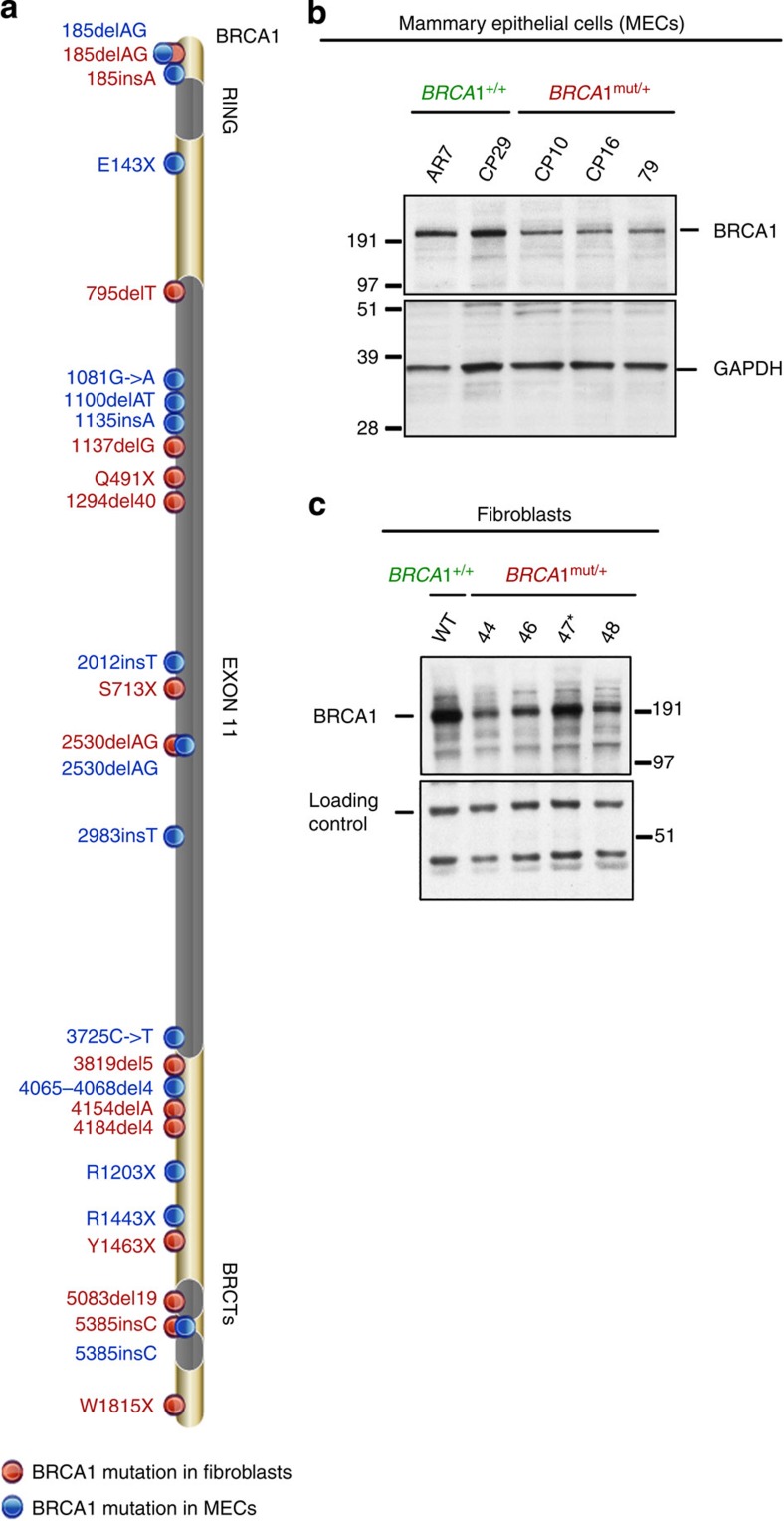
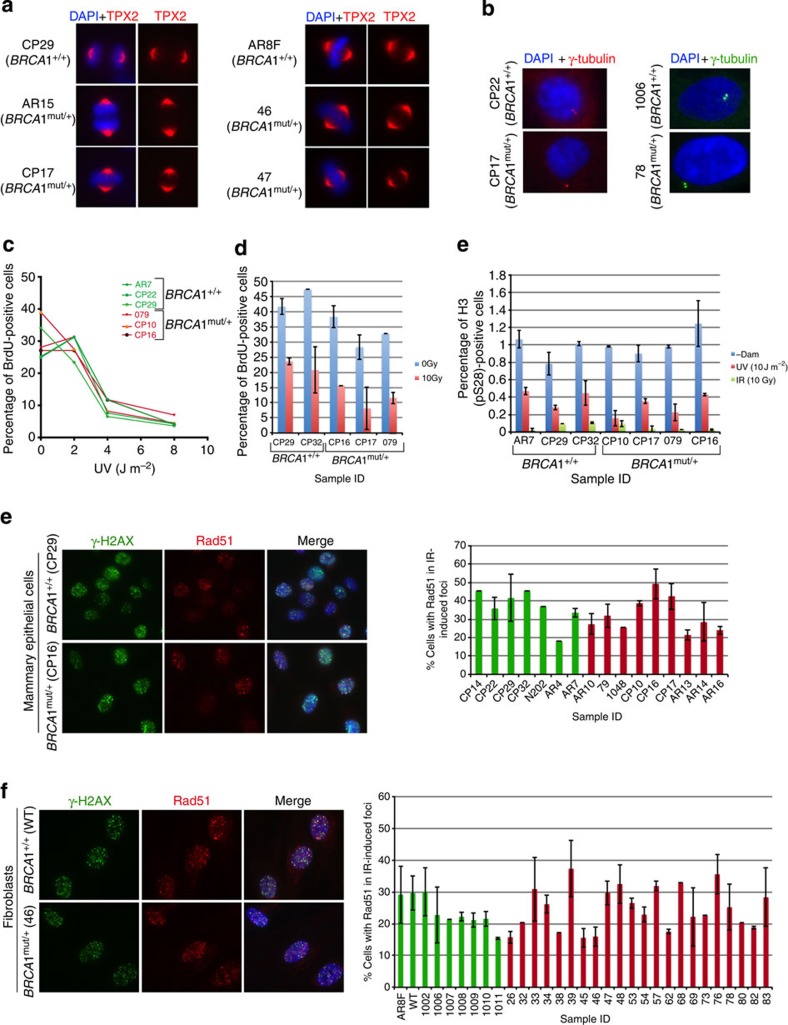
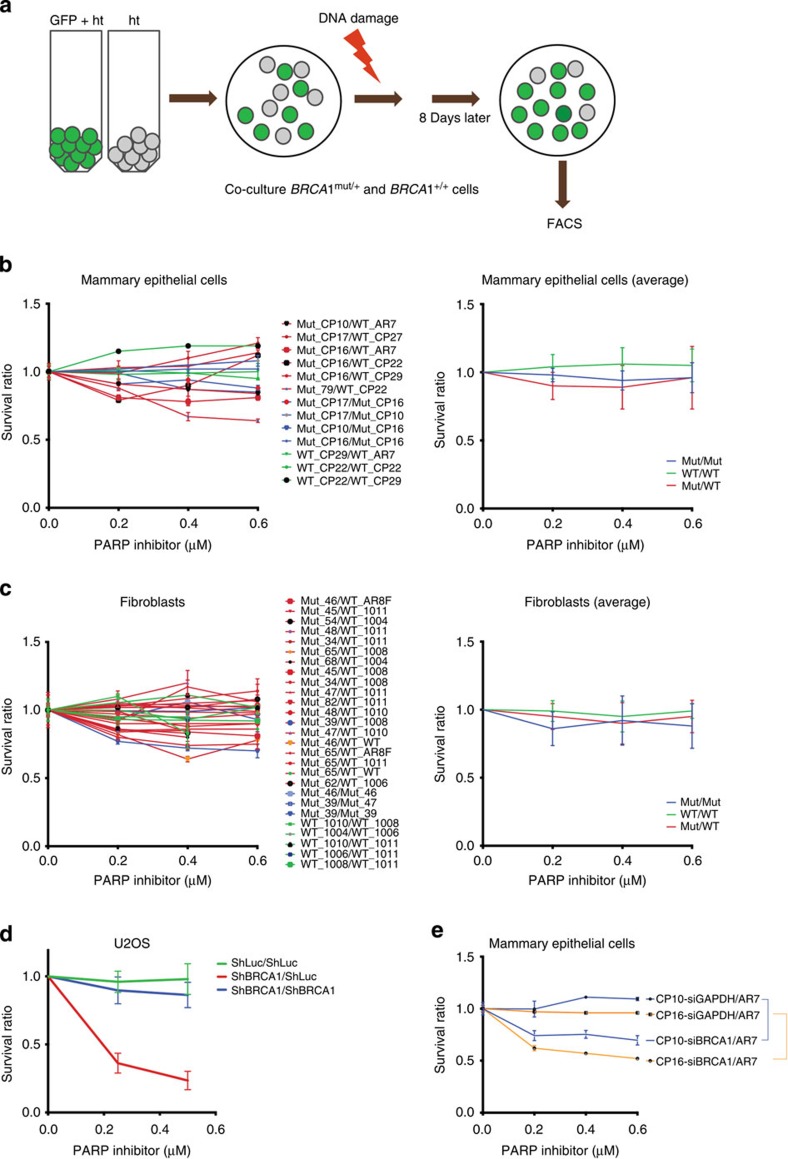
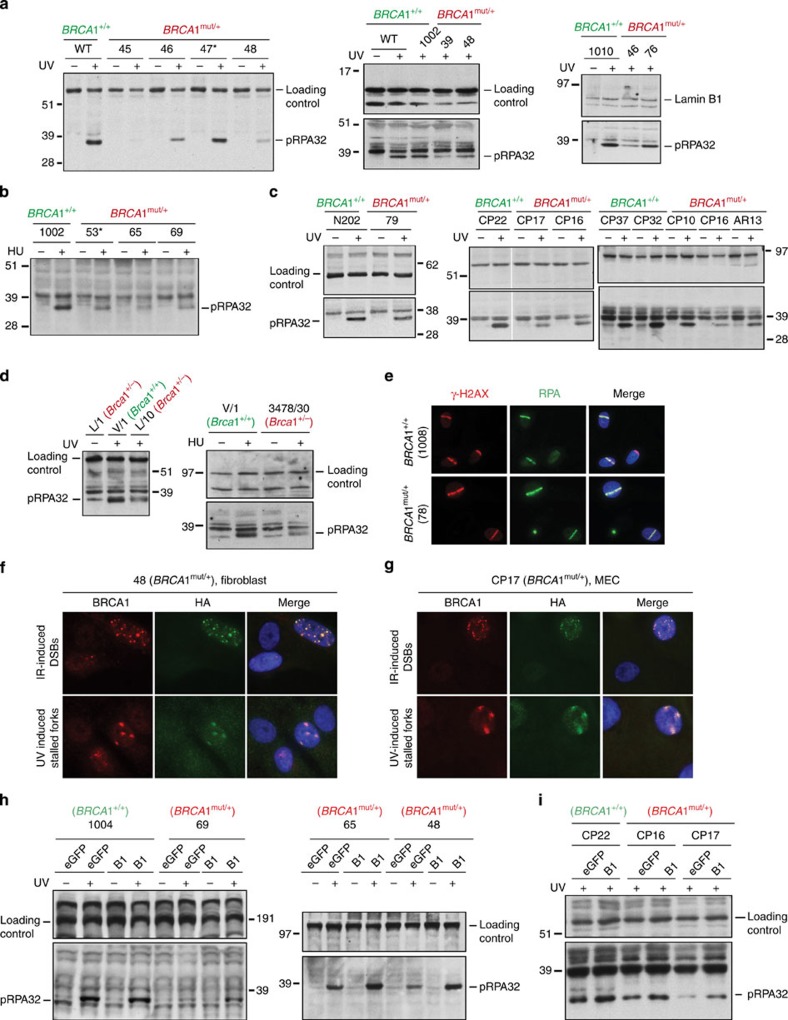
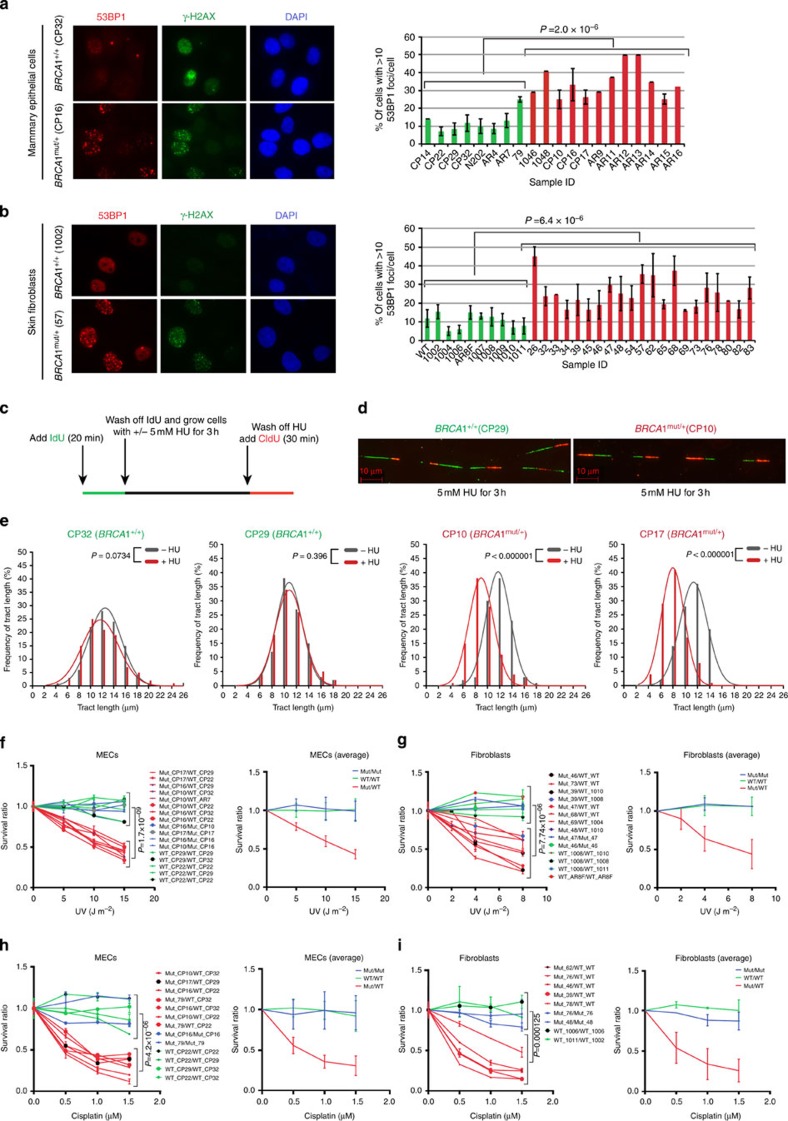
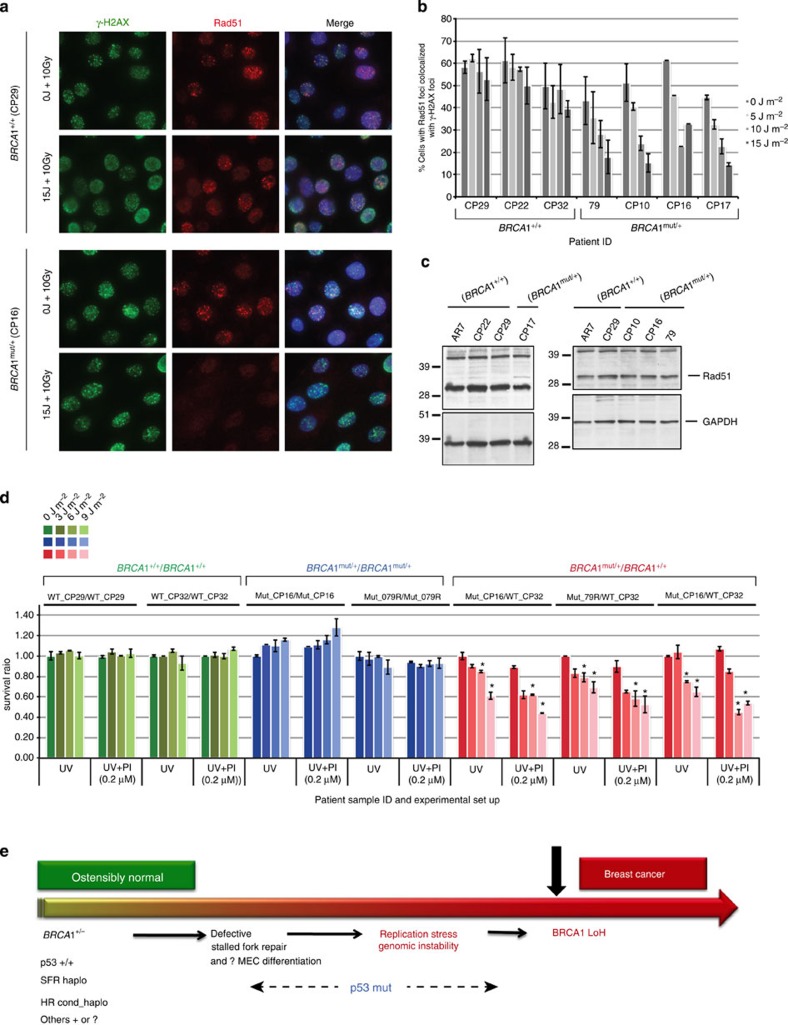
BRCA1-a breast and ovarian cancer suppressor gene-promotes genome integrity. To study the functionality of BRCA1 in the heterozygous state, we established a collection of primary human BRCA1(+/+) and BRCA1(mut/+) mammary epithelial cells and fibroblasts. Here we report that all BRCA1(mut/+) cells exhibited multiple normal BRCA1 functions, including the support of homologous recombination- type double-strand break repair (HR-DSBR), checkpoint functions, centrosome number control, spindle pole formation, Slug expression and satellite RNA suppression. In contrast, the same cells were defective in stalled replication fork repair and/or suppression of fork collapse, that is, replication stress. These defects were rescued by reconstituting BRCA1(mut/+) cells with wt BRCA1. In addition, we observed 'conditional' haploinsufficiency for HR-DSBR in BRCA1(mut/+) cells in the face of replication stress. Given the importance of replication stress in epithelial cancer development and of an HR defect in breast cancer pathogenesis, both defects are candidate contributors to tumorigenesis in BRCA1-deficient mammary tissue.
Figures
References
-
- King M.-C., Marks J. H., Mandell J. B. & Group N.Y.B.C.S. Breast and ovarian cancer risks due to inherited mutations in BRCA1 and BRCA2. Science 302, 643–646 (2003). - PubMed
-
- Narod S. A. & Foulkes W. D. BRCA1 and BRCA2: 1994 and beyond. Nat. Rev. Cancer. 4, 665–676 (2004). - PubMed
-
- Walsh T. & King M.-C. Ten genes for inherited breast cancer. Cancer Cell 11, 103–105 (2007). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
