

Estrogen signaling in metabolic inflammation
- PMID: 25400333
- PMCID: PMC4226184
- DOI: 10.1155/2014/615917
Estrogen signaling in metabolic inflammation
Abstract
There is extensive evidence supporting the interference of inflammatory activation with metabolism. Obesity, mainly visceral obesity, is associated with a low-grade inflammatory state, triggered by metabolic surplus where specialized metabolic cells such as adipocytes activate cellular stress initiating and sustaining the inflammatory program. The increasing prevalence of obesity, resulting in increased cardiometabolic risk and precipitating illness such as cardiovascular disease, type 2 diabetes, fatty liver, cirrhosis, and certain types of cancer, constitutes a good example of this association. The metabolic actions of estrogens have been studied extensively and there is also accumulating evidence that estrogens influence immune processes. However, the connection between these two fields of estrogen actions has been underacknowledged since little attention has been drawn towards the possible action of estrogens on the modulation of metabolism through their anti-inflammatory properties. In the present paper, we summarize knowledge on the modification inflammatory processes by estrogens with impact on metabolism and highlight major research questions on the field. Understanding the regulation of metabolic inflammation by estrogens may provide the basis for the development of therapeutic strategies to the management of metabolic dysfunctions.
Figures
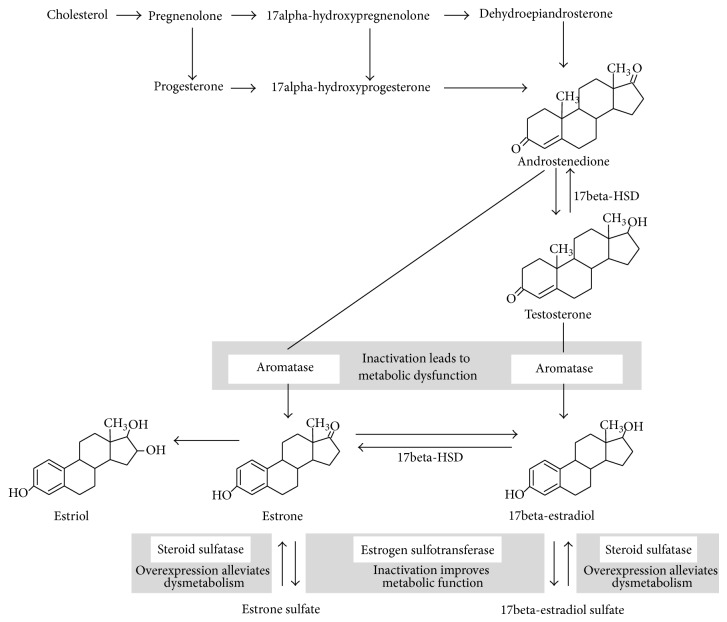
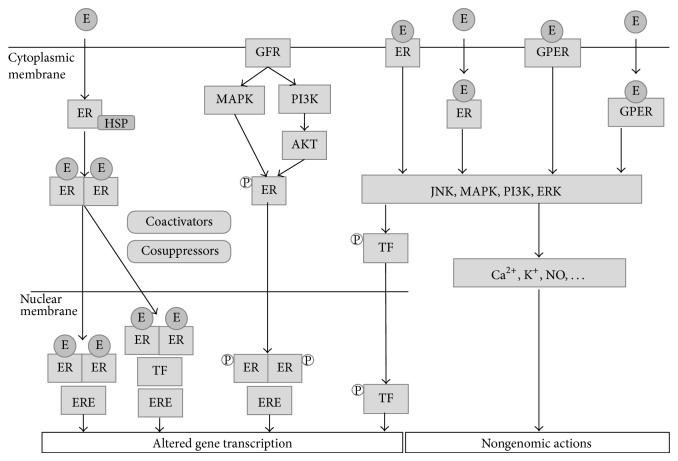
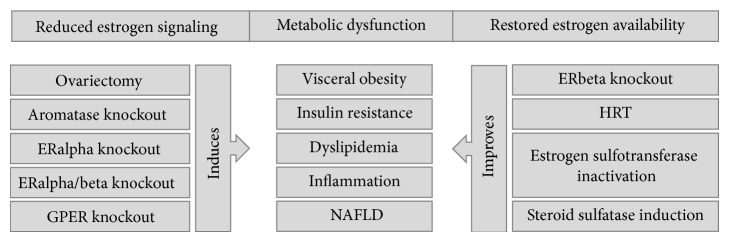
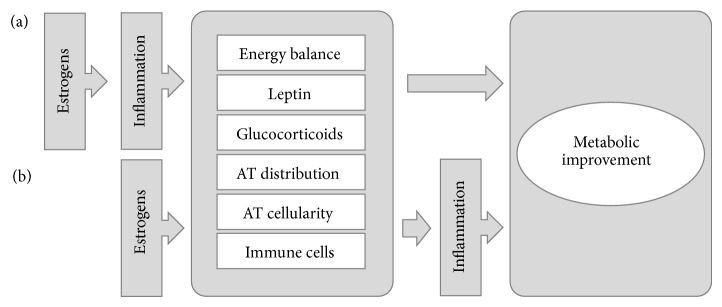
References
-
- Monteiro R. Chronic inflammation in the metabolic syndrome—emphasis on adipose tissue. In: Soares R., Costa C., editors. Oxidative Stress, Inflammation and Angiogenesis in the Metabolic Syndrome. London, UK: Springer; 2009. pp. 65–83.
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
