

Phylogeny and phylogeography of functional genes shared among seven terrestrial subsurface metagenomes reveal N-cycling and microbial evolutionary relationships
- PMID: 25400621
- PMCID: PMC4215791
- DOI: 10.3389/fmicb.2014.00531
Phylogeny and phylogeography of functional genes shared among seven terrestrial subsurface metagenomes reveal N-cycling and microbial evolutionary relationships
Abstract
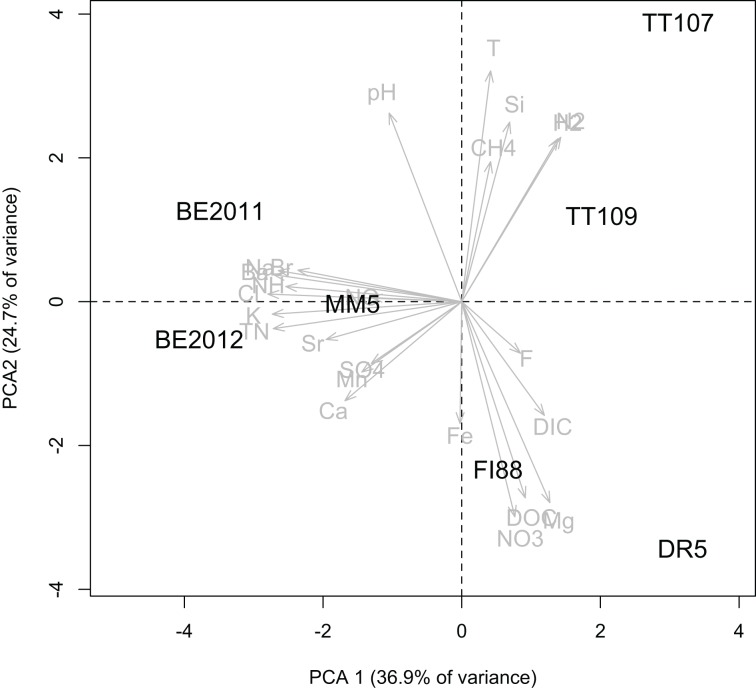
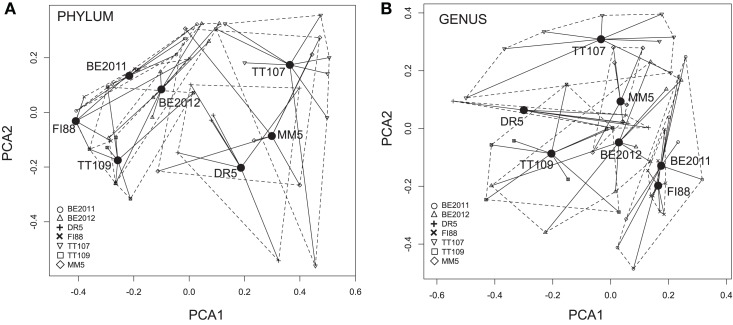
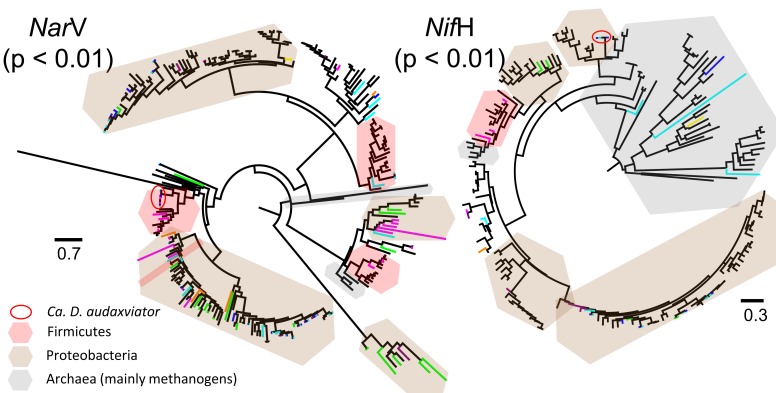
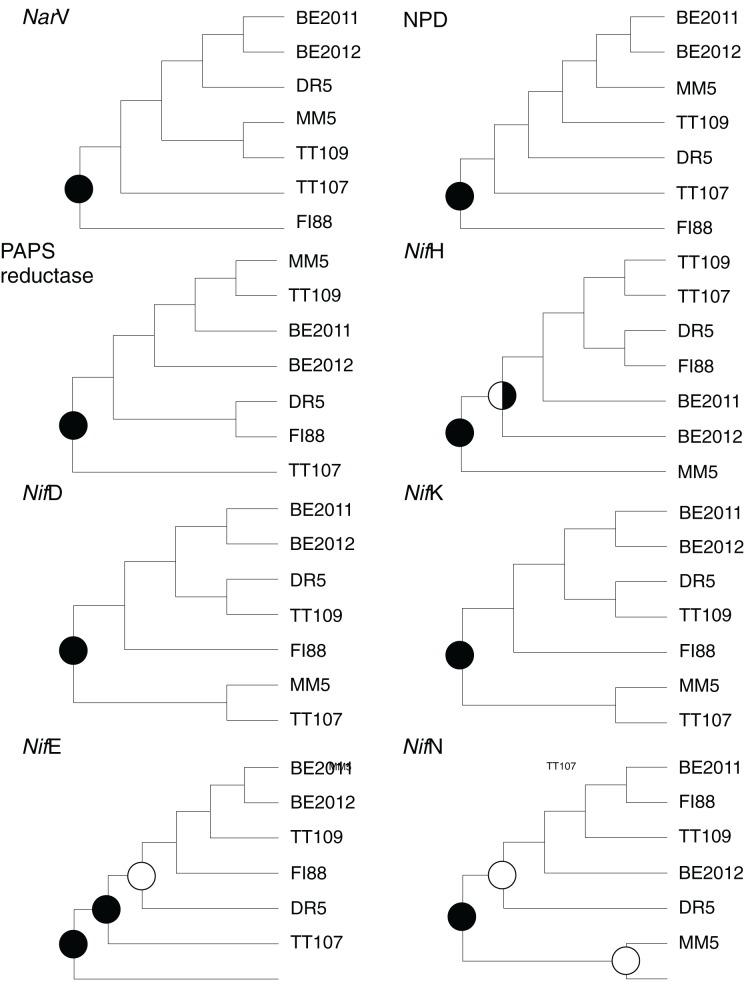
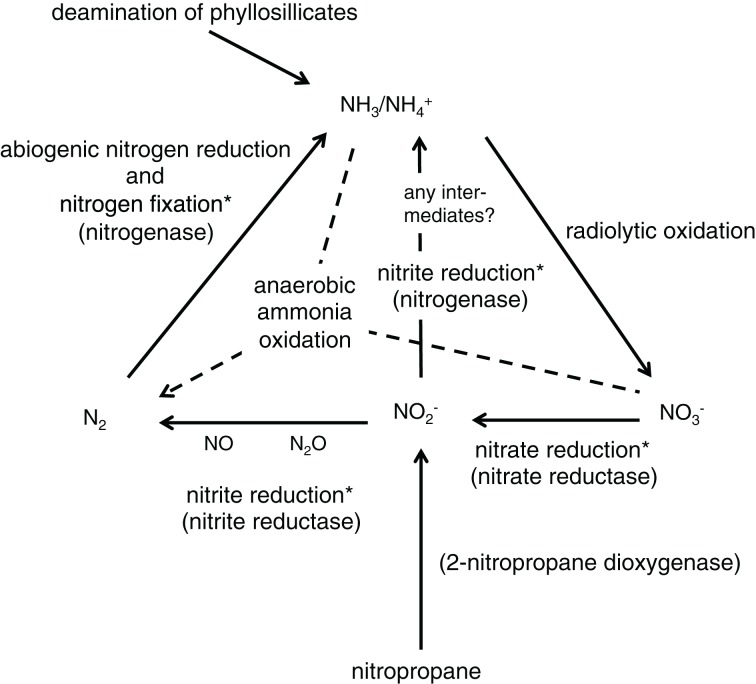
Comparative studies on community phylogenetics and phylogeography of microorganisms living in extreme environments are rare. Terrestrial subsurface habitats are valuable for studying microbial biogeographical patterns due to their isolation and the restricted dispersal mechanisms. Since the taxonomic identity of a microorganism does not always correspond well with its functional role in a particular community, the use of taxonomic assignments or patterns may give limited inference on how microbial functions are affected by historical, geographical and environmental factors. With seven metagenomic libraries generated from fracture water samples collected from five South African mines, this study was carried out to (1) screen for ubiquitous functions or pathways of biogeochemical cycling of CH4, S, and N; (2) to characterize the biodiversity represented by the common functional genes; (3) to investigate the subsurface biogeography as revealed by this subset of genes; and (4) to explore the possibility of using metagenomic data for evolutionary study. The ubiquitous functional genes are NarV, NPD, PAPS reductase, NifH, NifD, NifK, NifE, and NifN genes. Although these eight common functional genes were taxonomically and phylogenetically diverse and distinct from each other, the dissimilarity between samples did not correlate strongly with geographical or environmental parameters or residence time of the water. Por genes homologous to those of Thermodesulfovibrio yellowstonii detected in all metagenomes were deep lineages of Nitrospirae, suggesting that subsurface habitats have preserved ancestral genetic signatures that inform the study of the origin and evolution of prokaryotes.
Keywords: N-cycle; evolution; functional genes; phylogenetics; phylogeny; phylogeography; terrestrial subsurface.
Figures


References
-
- Anderson M. J., Walsh D. C. I. (2013). PERMANOVA, ANOSIM, and the Mantel test in the face of heterogeneous dispersions: what null hypothesis are you testing? Ecol. Monogr. 83, 557–574. 10.1890/12-2010.1 - DOI
-
- Andrews J. N., Wilson G. B. (1987). The composition of dissolved gases in deep groundwaters and groundwater degassing. Geol. Assoc. Can. Spec. Pap. 33, 245–252
-
- Baas Becking L. G. M. (1934). Geobiologie of Inleiding tot de Milieukunde. The Hague: W. P. Van Stockum & Zoon (in Dutch)
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases