

Harnessing Mechanistic Knowledge on Beneficial Versus Deleterious IFN-I Effects to Design Innovative Immunotherapies Targeting Cytokine Activity to Specific Cell Types
- PMID: 25400632
- PMCID: PMC4214202
- DOI: 10.3389/fimmu.2014.00526
Harnessing Mechanistic Knowledge on Beneficial Versus Deleterious IFN-I Effects to Design Innovative Immunotherapies Targeting Cytokine Activity to Specific Cell Types
Abstract
Type I interferons (IFN-I) were identified over 50 years ago as cytokines critical for host defense against viral infections. IFN-I promote anti-viral defense through two main mechanisms. First, IFN-I directly reinforce or induce de novo in potentially all cells the expression of effector molecules of intrinsic anti-viral immunity. Second, IFN-I orchestrate innate and adaptive anti-viral immunity. However, IFN-I responses can be deleterious for the host in a number of circumstances, including secondary bacterial or fungal infections, several autoimmune diseases, and, paradoxically, certain chronic viral infections. We will review the proposed nature of protective versus deleterious IFN-I responses in selected diseases. Emphasis will be put on the potentially deleterious functions of IFN-I in human immunodeficiency virus type 1 (HIV-1) infection, and on the respective roles of IFN-I and IFN-III in promoting resolution of hepatitis C virus (HCV) infection. We will then discuss how the balance between beneficial versus deleterious IFN-I responses is modulated by several key parameters including (i) the subtypes and dose of IFN-I produced, (ii) the cell types affected by IFN-I, and (iii) the source and timing of IFN-I production. Finally, we will speculate how integration of this knowledge combined with advanced biochemical manipulation of the activity of the cytokines should allow designing innovative immunotherapeutic treatments in patients. Specifically, we will discuss how induction or blockade of specific IFN-I responses in targeted cell types could promote the beneficial functions of IFN-I and/or dampen their deleterious effects, in a manner adapted to each disease.
Keywords: bioengineering; chronic viral infections; dendritic cells; immunotherapy; type I interferons.
Figures
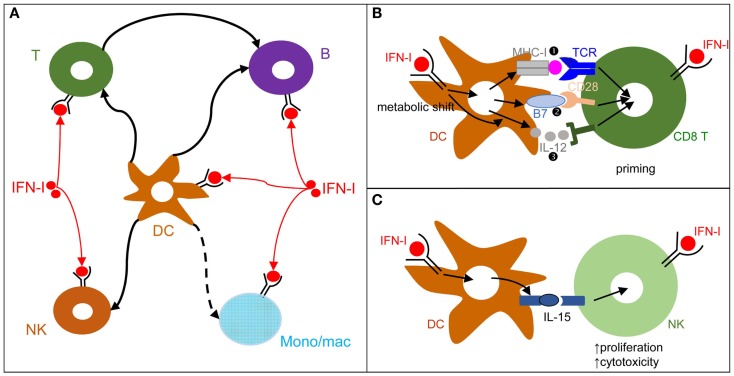
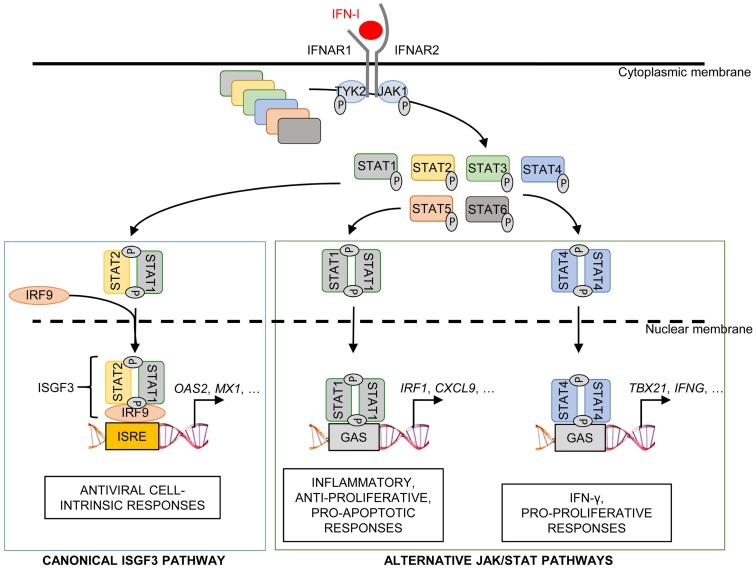
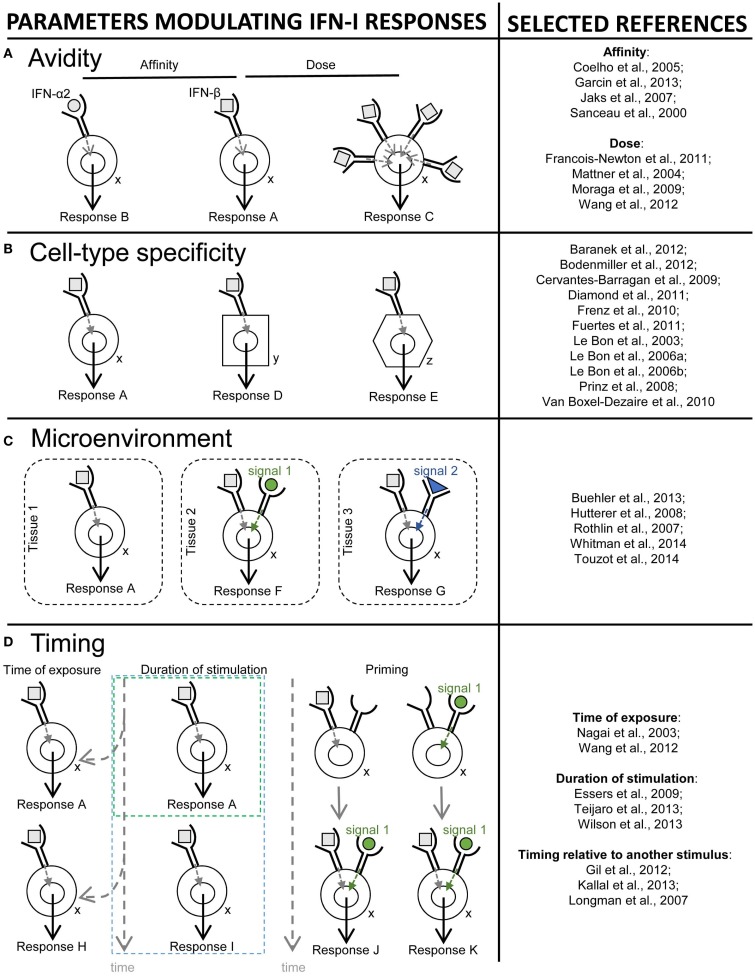
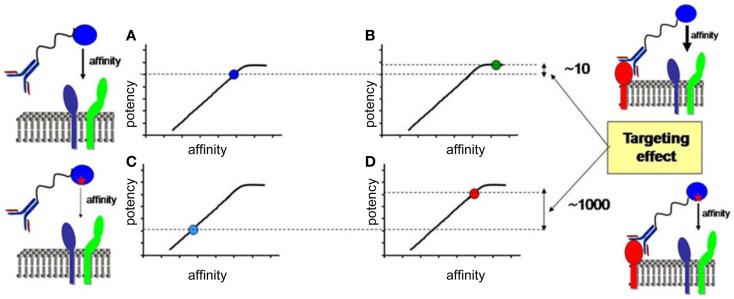
 ), co-stimulation (
), co-stimulation ( ), and cytokines (
), and cytokines ( ). This depends both on IFN-I-dependent transcriptional induction in DC of some of the corresponding genes and on IFN-I-dependent metabolic reprogramming of DC. (C) DC cell-intrinsic responses to IFN-I endow them to deliver appropriate signals, in particular IL-15 trans-presentation, for NK cell activation. See main text for further details.
). This depends both on IFN-I-dependent transcriptional induction in DC of some of the corresponding genes and on IFN-I-dependent metabolic reprogramming of DC. (C) DC cell-intrinsic responses to IFN-I endow them to deliver appropriate signals, in particular IL-15 trans-presentation, for NK cell activation. See main text for further details.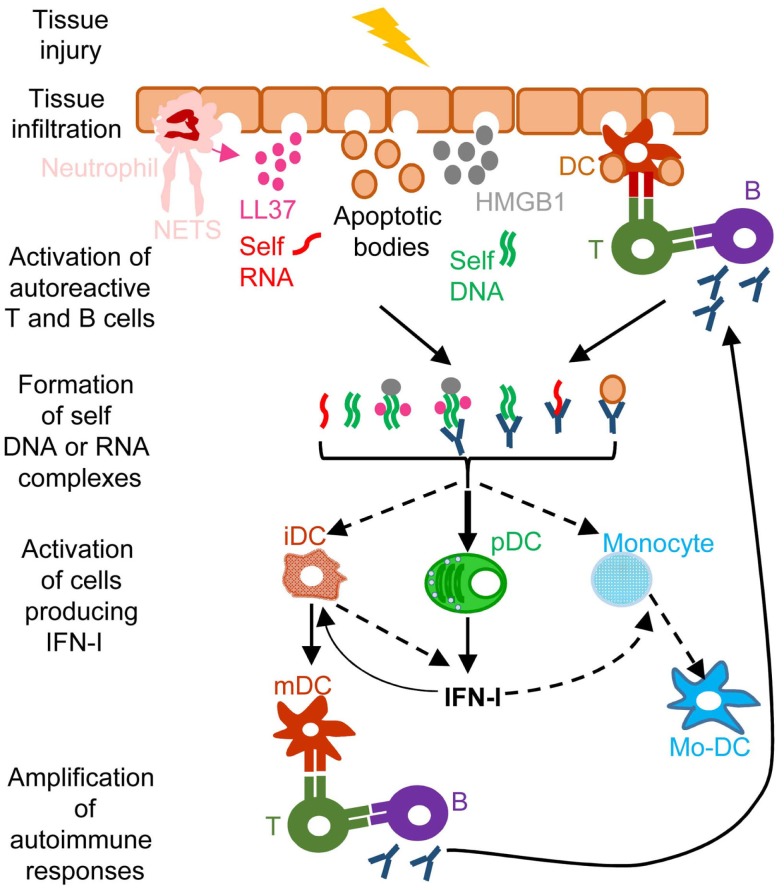
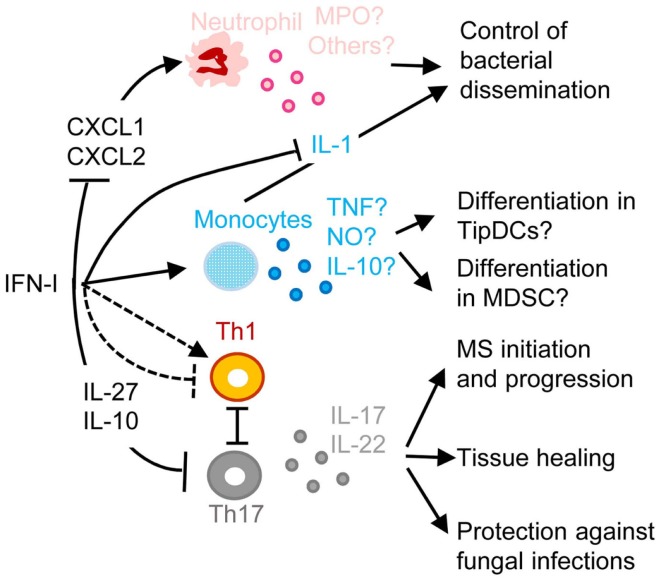
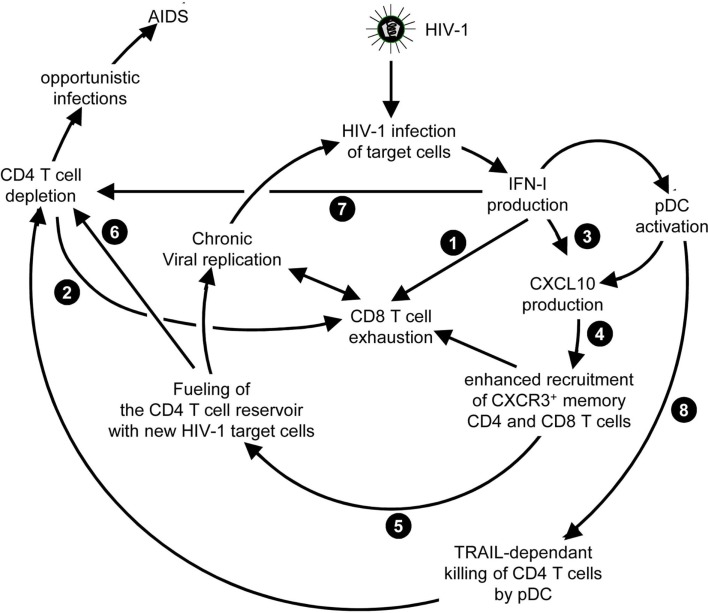 ) and indirect () promotion of the exhaustion of anti-viral CD8 T cell responses, as well as direct (
) and indirect () promotion of the exhaustion of anti-viral CD8 T cell responses, as well as direct ( ) and indirect (-to-
) and indirect (-to- , and
, and  ) promotion of CD4 T cell depletion with a proposed central role of pDC in this deleterious process. Altogether, these mechanisms may sustain a vicious circle of reciprocal activation between chronic viral replication and deleterious immune responses, driving the progressive depletion of all CD4 T cells ultimately causing the enhanced susceptibility to opportunistic infections characteristic of the acquired immunodeficiency syndrome (AIDS). See main text for further details.
) promotion of CD4 T cell depletion with a proposed central role of pDC in this deleterious process. Altogether, these mechanisms may sustain a vicious circle of reciprocal activation between chronic viral replication and deleterious immune responses, driving the progressive depletion of all CD4 T cells ultimately causing the enhanced susceptibility to opportunistic infections characteristic of the acquired immunodeficiency syndrome (AIDS). See main text for further details.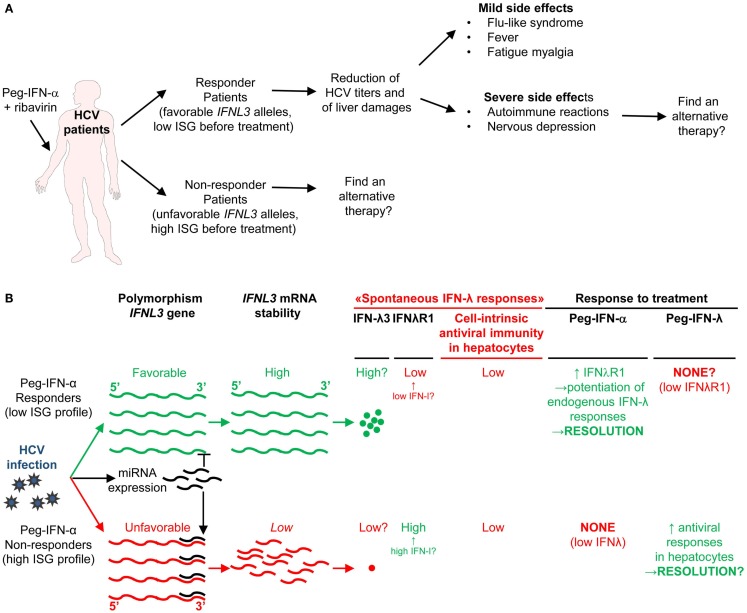
References
-
- Zhang SY, Boisson-Dupuis S, Chapgier A, Yang K, Bustamante J, Puel A, et al. Inborn errors of interferon (IFN)-mediated immunity in humans: insights into the respective roles of IFN-alpha/beta, IFN-gamma, and IFN-lambda in host defense. Immunol Rev (2008) 226:29–40.10.1111/j.1600-065X.2008.00698.x - DOI - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources