

Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice
- PMID: 25402683
- PMCID: PMC4894846
- DOI: 10.1038/ncb3064
Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice
Abstract
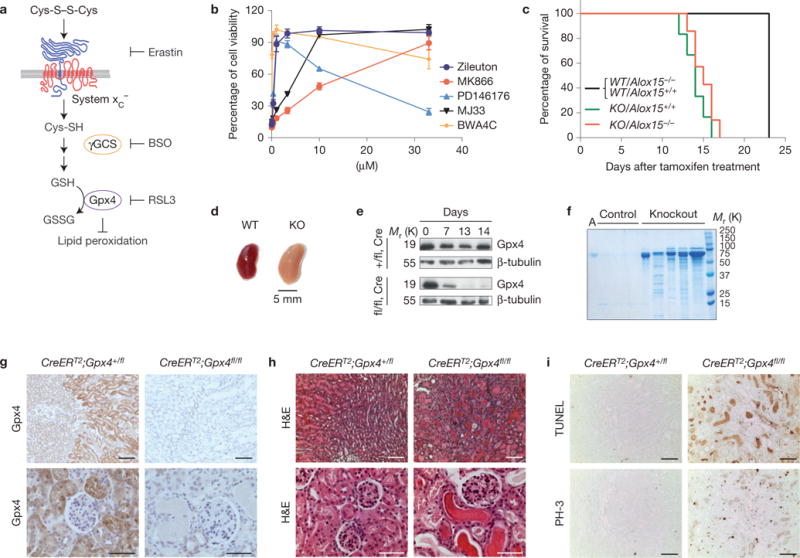
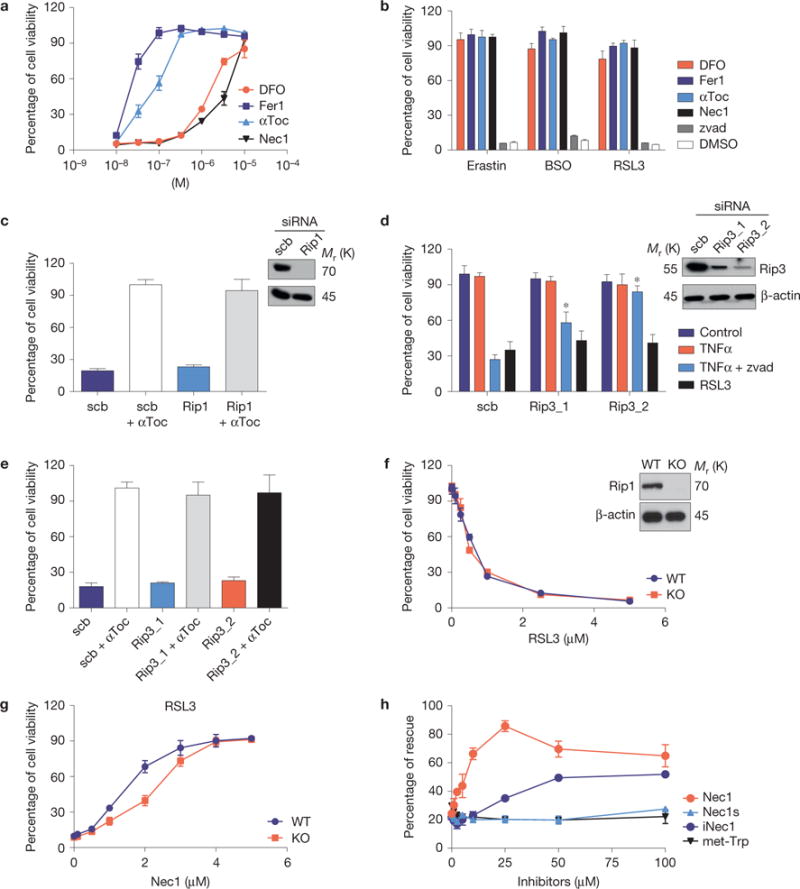
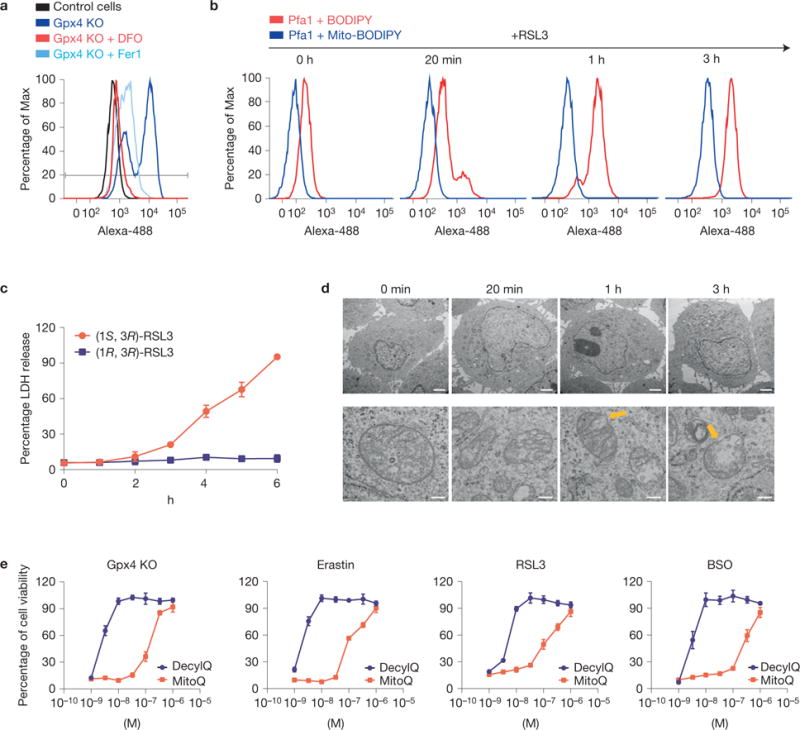
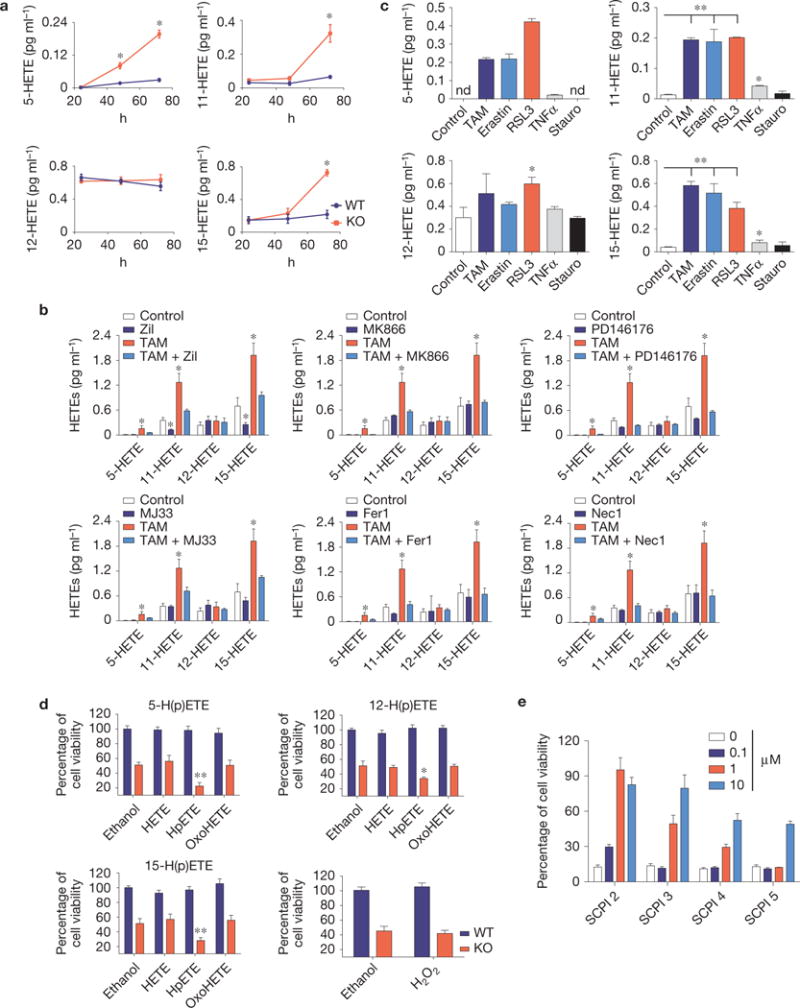
Ferroptosis is a non-apoptotic form of cell death induced by small molecules in specific tumour types, and in engineered cells overexpressing oncogenic RAS. Yet, its relevance in non-transformed cells and tissues is unexplored and remains enigmatic. Here, we provide direct genetic evidence that the knockout of glutathione peroxidase 4 (Gpx4) causes cell death in a pathologically relevant form of ferroptosis. Using inducible Gpx4(-/-) mice, we elucidate an essential role for the glutathione/Gpx4 axis in preventing lipid-oxidation-induced acute renal failure and associated death. We furthermore systematically evaluated a library of small molecules for possible ferroptosis inhibitors, leading to the discovery of a potent spiroquinoxalinamine derivative called Liproxstatin-1, which is able to suppress ferroptosis in cells, in Gpx4(-/-) mice, and in a pre-clinical model of ischaemia/reperfusion-induced hepatic damage. In sum, we demonstrate that ferroptosis is a pervasive and dynamic form of cell death, which, when impeded, promises substantial cytoprotection.
Conflict of interest statement
The authors declare that a patent application has been filed for some of the compounds described in this work.
Figures
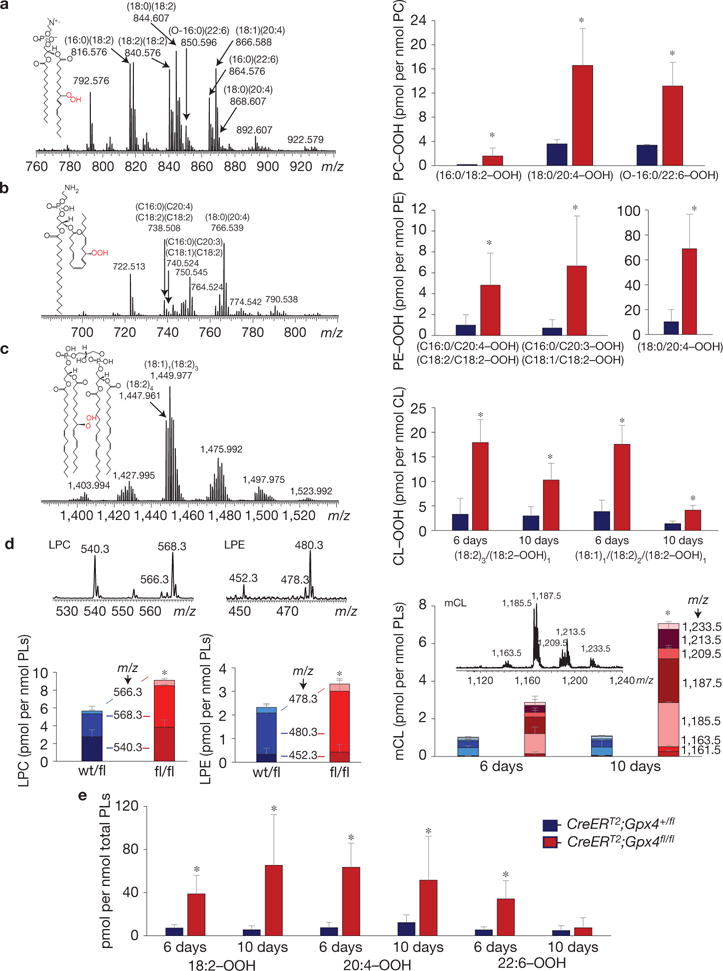
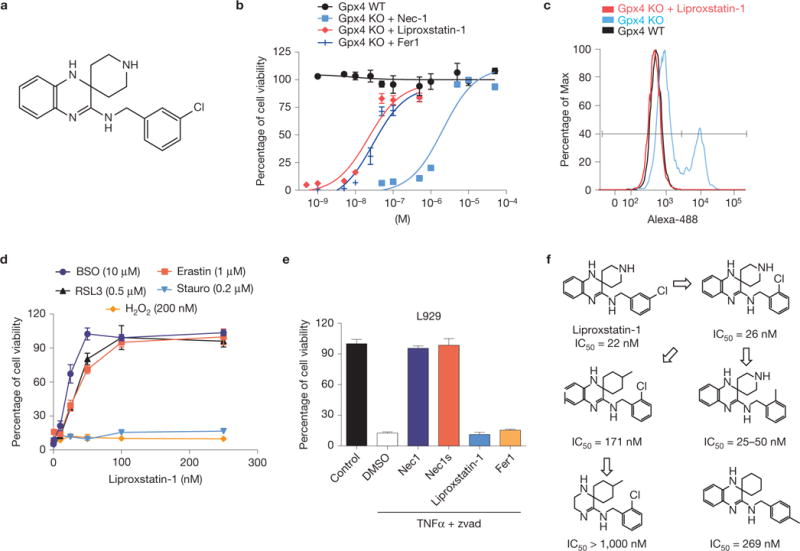
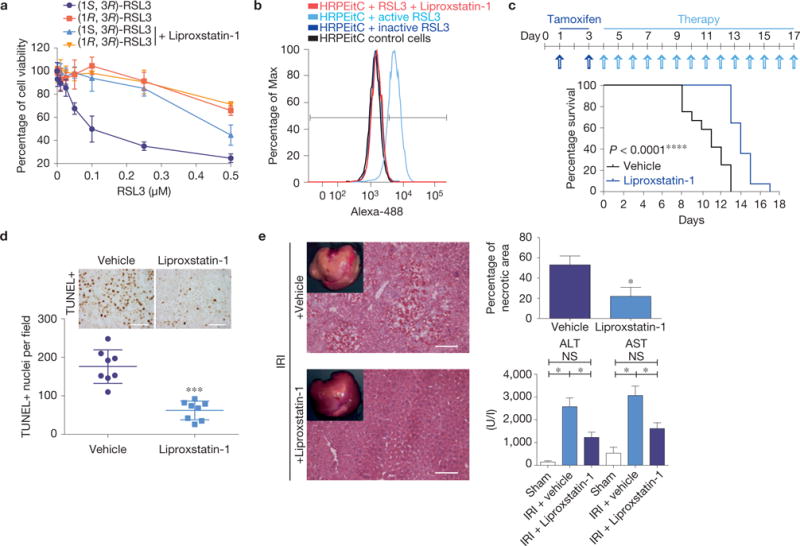
References
-
- Ofengeim D, Yuan J. Regulation of RIP1 kinase signalling at the crossroads of inflammation and cell death. Nat Rev Mol Cell Biol. 2013;14:727–736. - PubMed
-
- Fulda S, Vucic D. Targeting IAP proteins for therapeutic intervention in cancer. Nat Rev Drug Discov. 2012;11:109–124. - PubMed
-
- Vanden Berghe T, Linkermann A, Jouan-Lanhouet S, Walczak H, Vandenabeele P. Regulated necrosis: the expanding network of non-apoptotic cell death pathways. Nat Rev Mol Cell Biol. 2014;15:135–147. - PubMed
-
- Vandenabeele P, Galluzzi L, Vanden Berghe T, Kroemer G. Molecular mechanisms of necroptosis: an ordered cellular explosion. Nat Rev Mol Cell Biol. 2010;11:700–714. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
