

Foxk proteins repress the initiation of starvation-induced atrophy and autophagy programs
- PMID: 25402684
- PMCID: PMC4250422
- DOI: 10.1038/ncb3062
Foxk proteins repress the initiation of starvation-induced atrophy and autophagy programs
Abstract
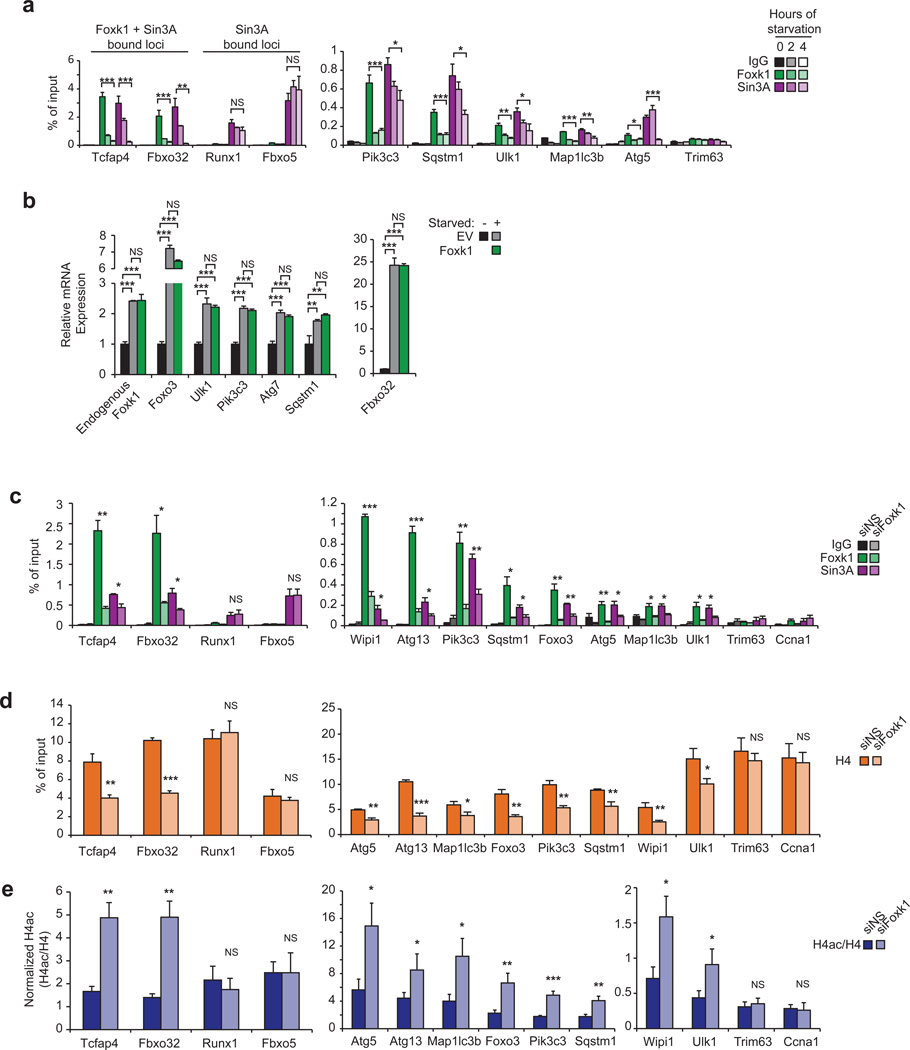
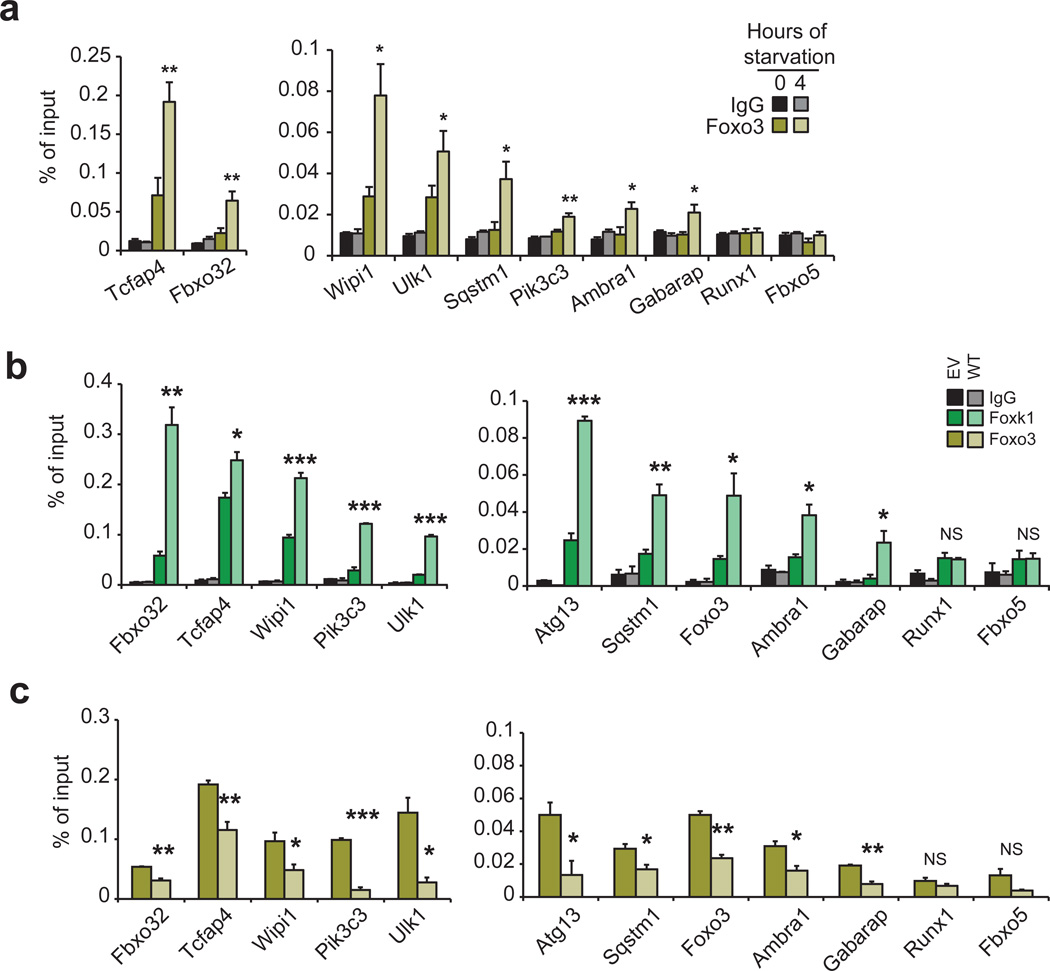
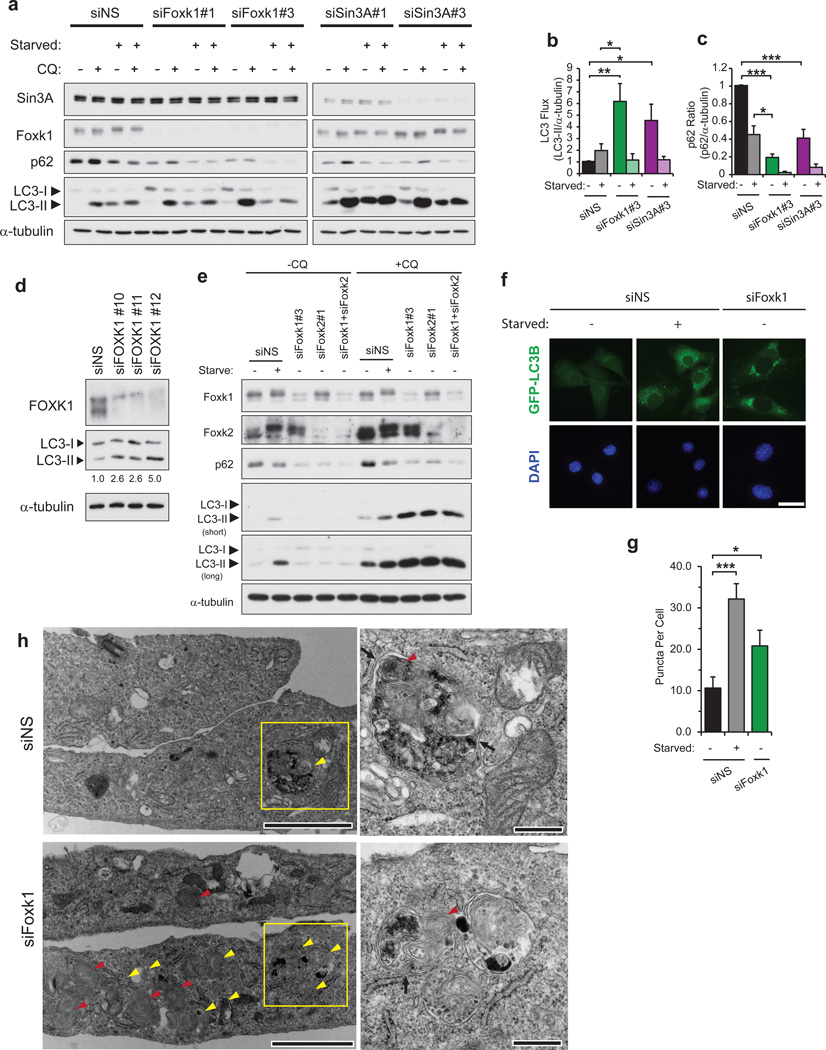
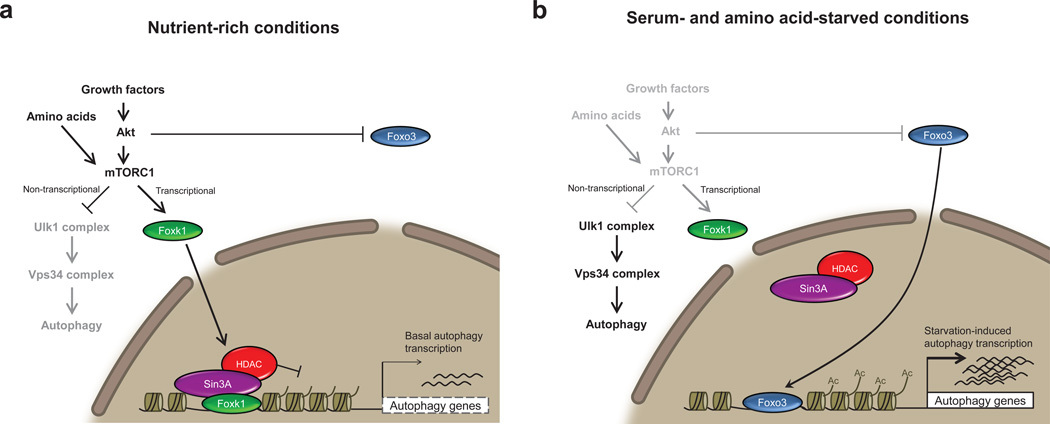
Autophagy is the primary catabolic process triggered in response to starvation. Although autophagic regulation within the cytosolic compartment is well established, it is becoming clear that nuclear events also regulate the induction or repression of autophagy. Nevertheless, a thorough understanding of the mechanisms by which sequence-specific transcription factors modulate expression of genes required for autophagy is lacking. Here, we identify Foxk proteins (Foxk1 and Foxk2) as transcriptional repressors of autophagy in muscle cells and fibroblasts. Interestingly, Foxk1/2 serve to counter-balance another forkhead transcription factor, Foxo3, which induces an overlapping set of autophagic and atrophic targets in muscle. Foxk1/2 specifically recruits Sin3A-HDAC complexes to restrict acetylation of histone H4 and expression of critical autophagy genes. Remarkably, mTOR promotes the transcriptional activity of Foxk1 by facilitating nuclear entry to specifically limit basal levels of autophagy in nutrient-rich conditions. Our study highlights an ancient, conserved mechanism whereby nutritional status is interpreted by mTOR to restrict autophagy by repressing essential autophagy genes through Foxk-Sin3-mediated transcriptional control.
Figures
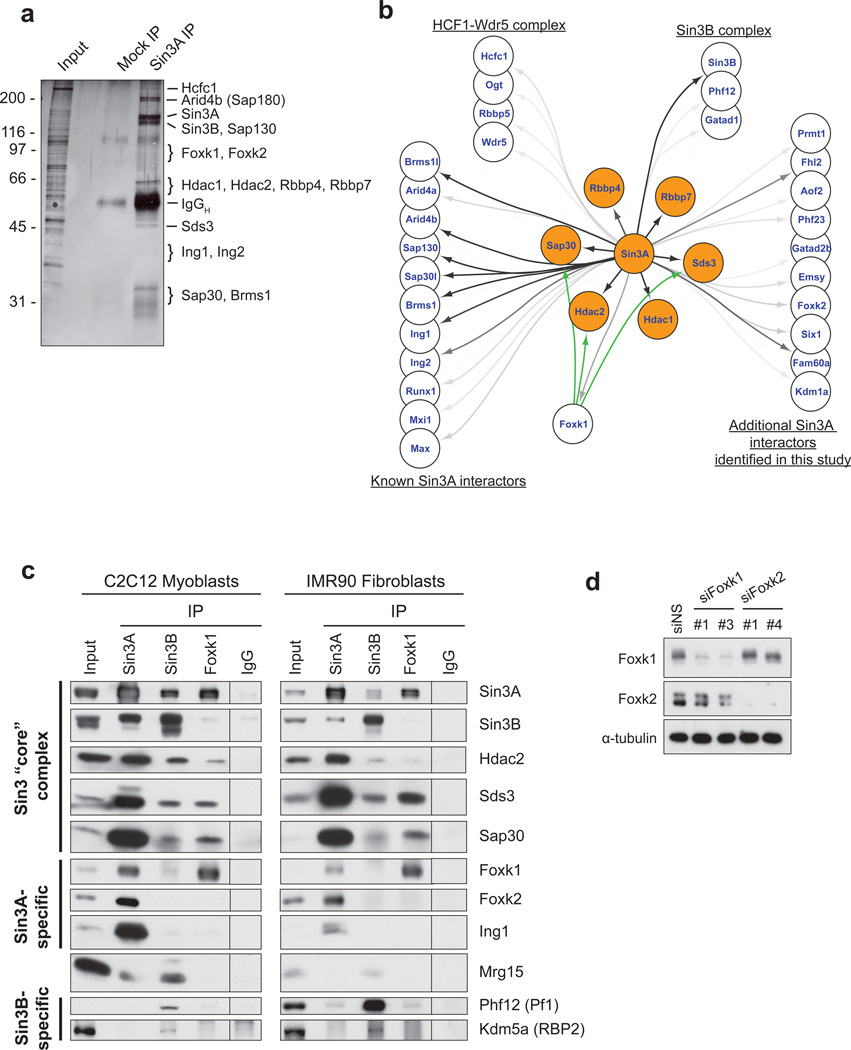
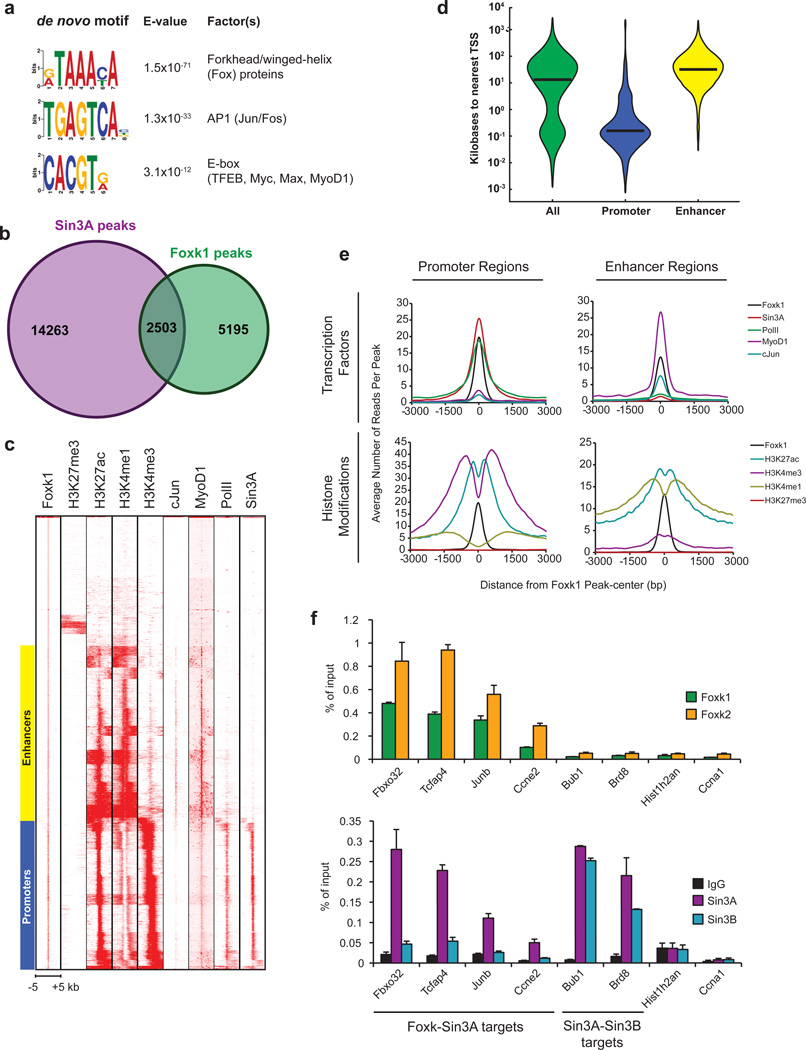
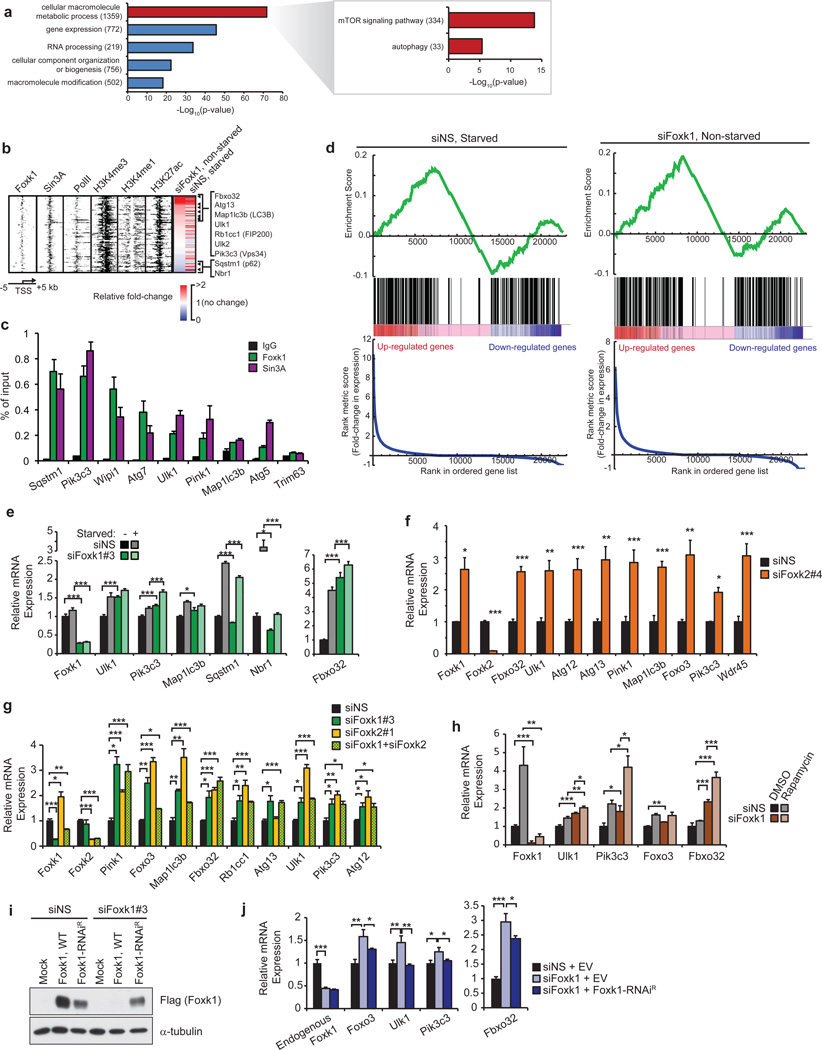
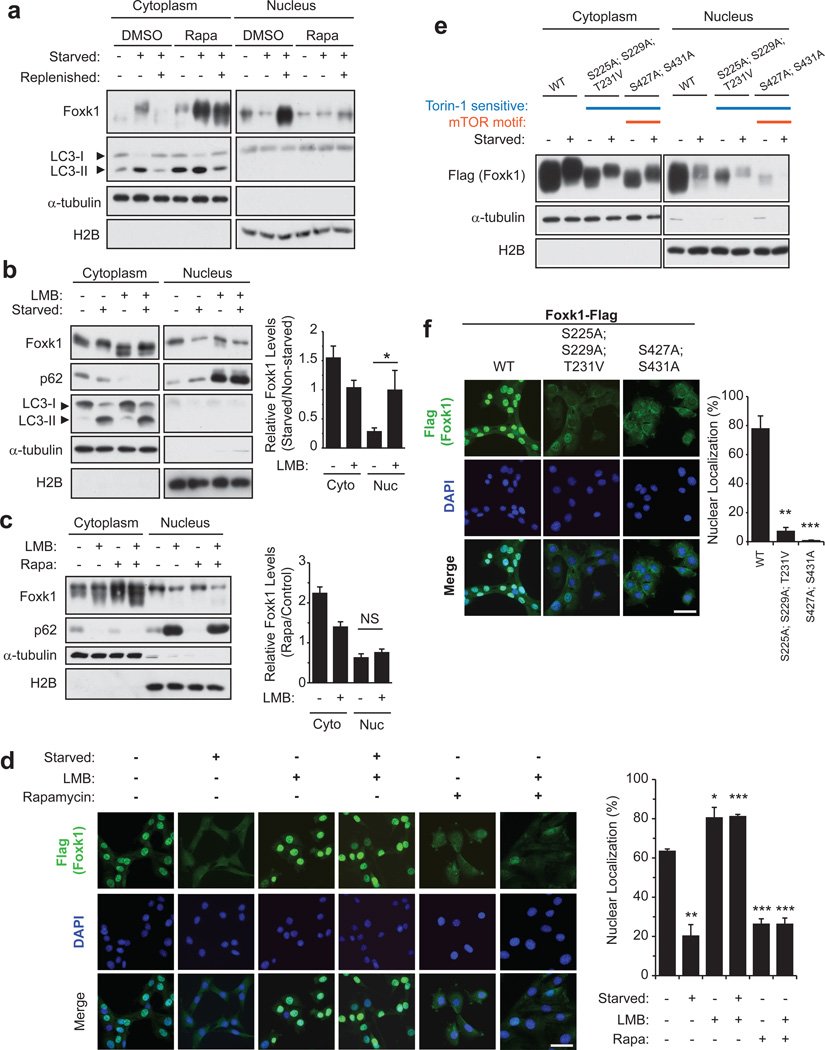
References
-
- Mizushima N. Autophagy: process and function. Genes Dev. 2007;21:2861–2873. - PubMed
-
- Füllgrabe J, Klionsky DJ, Joseph B. The return of the nucleus: transcriptional and epigenetic control of autophagy. Nat. Rev. Mol. Cell Biol. 2014;15:65–74. - PubMed
-
- Pietrocola F, et al. Regulation of autophagy by stress-responsive transcription factors. Semin. Cancer Biol. 2013;23:310–322. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
