

Increased SHP-1 protein expression by high glucose levels reduces nephrin phosphorylation in podocytes
- PMID: 25404734
- PMCID: PMC4281738
- DOI: 10.1074/jbc.M114.612721
Increased SHP-1 protein expression by high glucose levels reduces nephrin phosphorylation in podocytes
Abstract
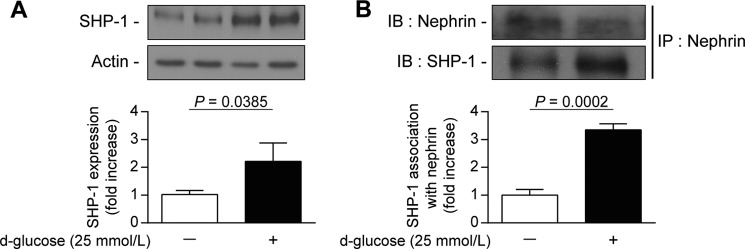
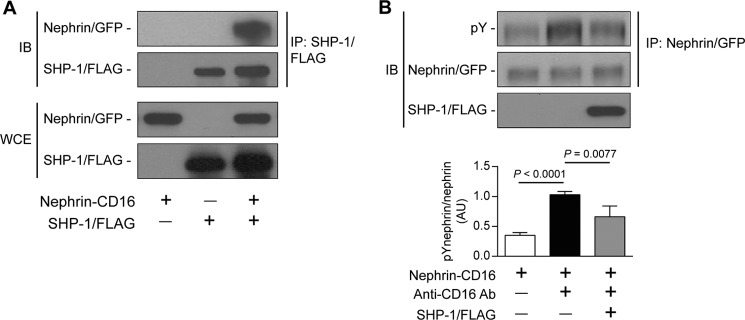
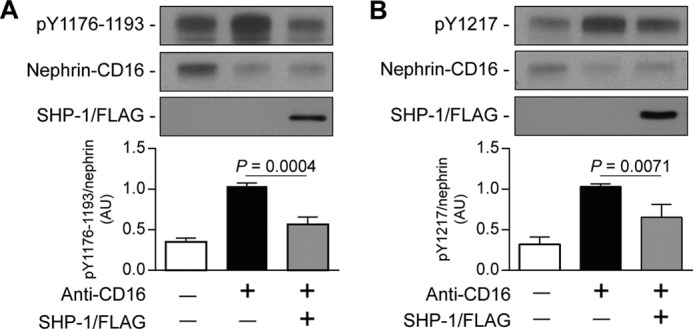
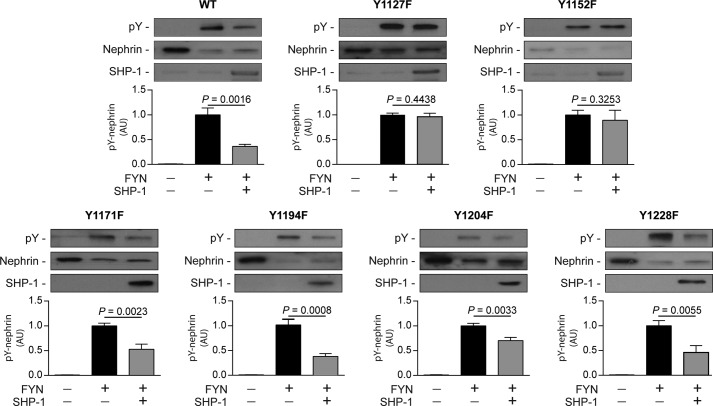
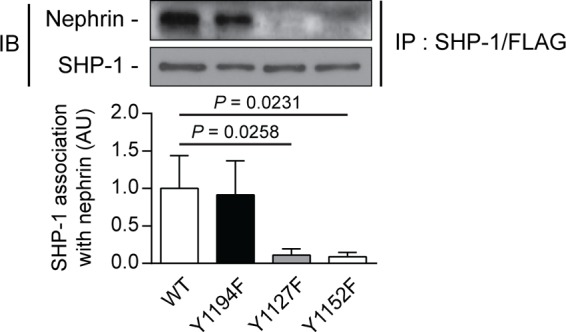
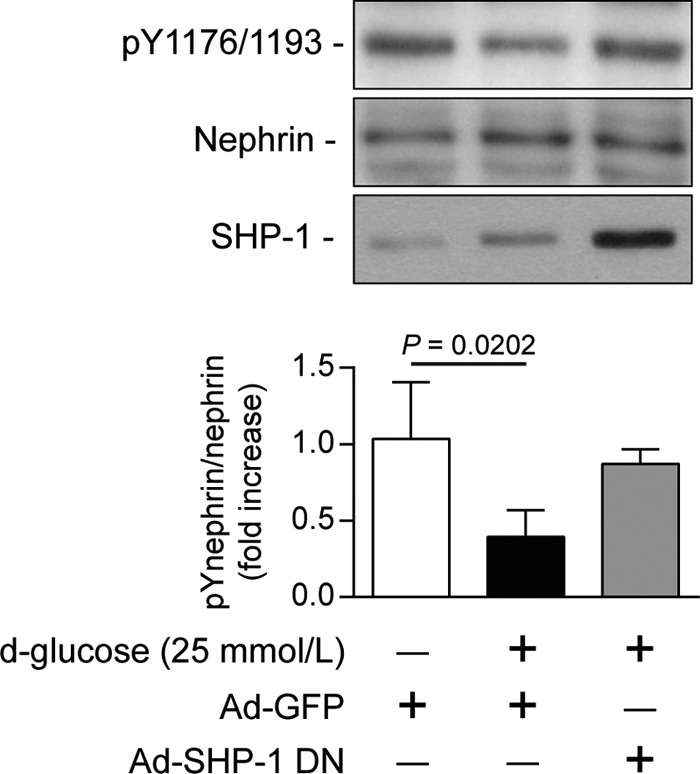
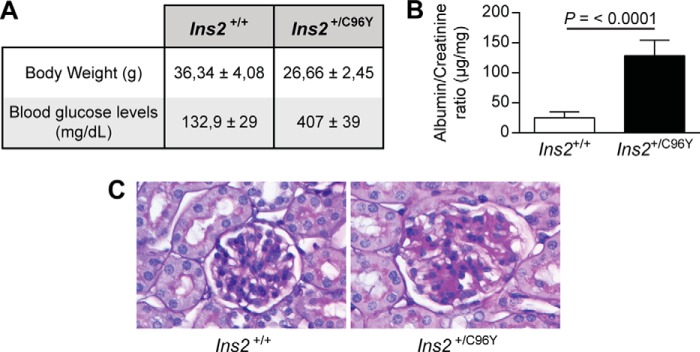
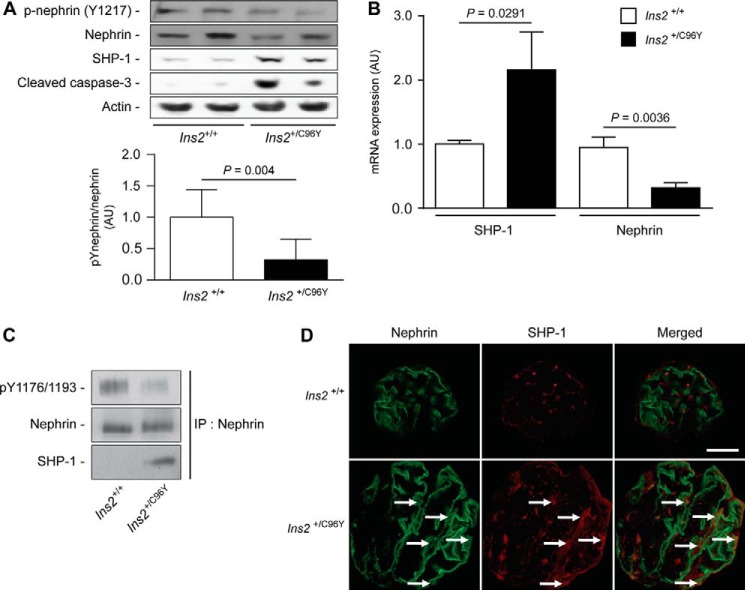
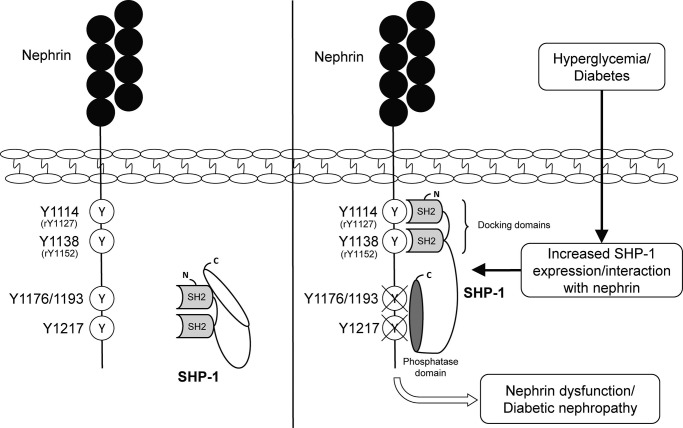
Nephrin, a critical podocyte membrane component that is reduced in diabetic nephropathy, has been shown to activate phosphotyrosine signaling pathways in human podocytes. Nephrin signaling is important to reduce cell death induced by apoptotic stimuli. We have shown previously that high glucose level exposure and diabetes increased the expression of SHP-1, causing podocyte apoptosis. SHP-1 possesses two Src homology 2 domains that serve as docking elements to dephosphorylate tyrosine residues of target proteins. However, it remains unknown whether SHP-1 interacts with nephrin and whether its elevated expression affects the nephrin phosphorylation state in diabetes. Here we show that human podocytes exposed to high glucose levels exhibited elevated expression of SHP-1, which was associated with nephrin. Coexpression of nephrin-CD16 and SHP-1 reduced nephrin tyrosine phosphorylation in transfected human embryonic kidney 293 cells. A single tyrosine-to-phenylalanine mutation revealed that rat nephrin Tyr(1127) and Tyr(1152) are required to allow SHP-1 interaction with nephrin. Overexpression of dominant negative SHP-1 in human podocytes prevented high glucose-induced reduction of nephrin phosphorylation. In vivo, immunoblot analysis demonstrated that nephrin expression and phosphorylation were decreased in glomeruli of type 1 diabetic Akita mice (Ins2(+/C96Y)) compared with control littermate mice (Ins2(+/+)), and this was associated with elevated SHP-1 and cleaved caspase-3 expression. Furthermore, immunofluorescence analysis indicated increased colocalization of SHP-1 with nephrin in diabetic mice compared with control littermates. In conclusion, our results demonstrate that high glucose exposure increases SHP-1 interaction with nephrin, causing decreased nephrin phosphorylation, which may, in turn, contribute to diabetic nephropathy.
Keywords: Diabetes; Nephrin; Nephrology; Phosphotyrosine; Podocyte; Protein-Tyrosine Phosphatase (Tyrosine Phosphatase); Src Homology 2 domain (SH2 domain).
© 2015 by The American Society for Biochemistry and Molecular Biology, Inc.
Figures
References
-
- Kaplan J. M., Kim S. H., North K. N., Rennke H., Correia L. A., Tong H. Q., Mathis B. J., Rodríguez-Pérez J. C., Allen P. G., Beggs A. H., Pollak M. R. (2000) Mutations in ACTN4, encoding alpha-actinin-4, cause familial focal segmental glomerulosclerosis. Nat. Genet. 24, 251–256 - PubMed
-
- Collins A. J., Kasiske B., Herzog C., Chavers B., Foley R., Gilbertson D., Grimm R., Liu J., Louis T., Manning W., McBean M., Murray A., St Peter W., Xue J., Fan Q., Guo H., Li Q., Li S., Qiu Y., Li S., Roberts T., Skeans M., Snyder J., Solid C., Wang C., Weinhandl E., Zhang R., Arko C., Chen S.-C. C., Dalleska F., Daniels F., Dunning S., Ebben J., Frazier E., Hanzlik C., Johnson R., Sheets D., Wang X., Forrest B., Berrini D., Constantini E., Everson S., Eggers P., Agodoa L. (2006) Excerpts from the United States Renal Data System 2006 Annual Data Report. Am. J. Kidney diseases 49, S1–S296 - PubMed
-
- Sarnak M. J., Levey A. S., Schoolwerth A. C., Coresh J., Culleton B., Hamm L. L., McCullough P. A., Kasiske B. L., Kelepouris E., Klag M. J., Parfrey P., Pfeffer M., Raij L., Spinosa D. J., Wilson P. W., American Heart Association Councils on Kidney in Cardiovascular Disease, High Blood Pressure Research, Clinical Cardiology, and Epidemiology and Prevention. (2003) Kidney disease as a risk factor for development of cardiovascular disease: a statement from the American Heart Association Councils on Kidney in Cardiovascular Disease, High Blood Pressure Research, Clinical Cardiology, and Epidemiology and Prevention. Circulation 108, 2154–2169 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
