

Deficient Autophagy Results in Mitochondrial Dysfunction and FSGS
- PMID: 25406339
- PMCID: PMC4413752
- DOI: 10.1681/ASN.2013111202
Deficient Autophagy Results in Mitochondrial Dysfunction and FSGS
Abstract
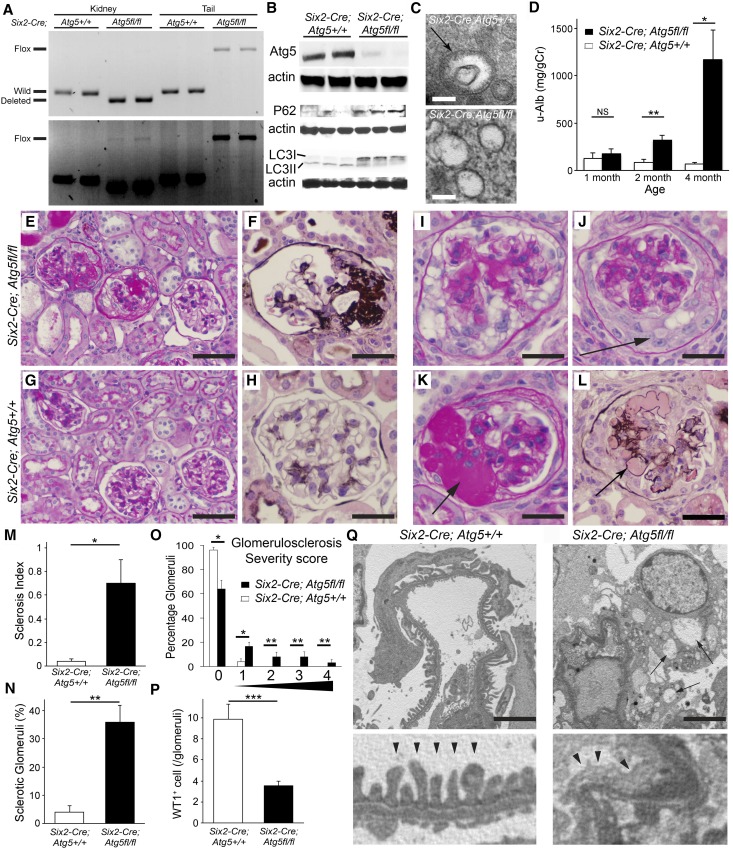
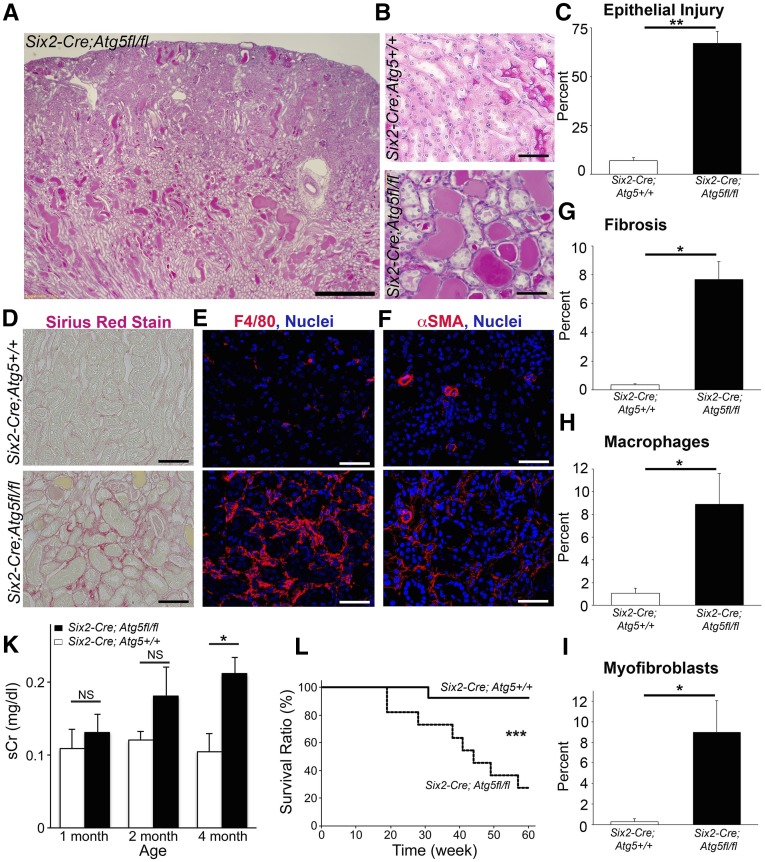
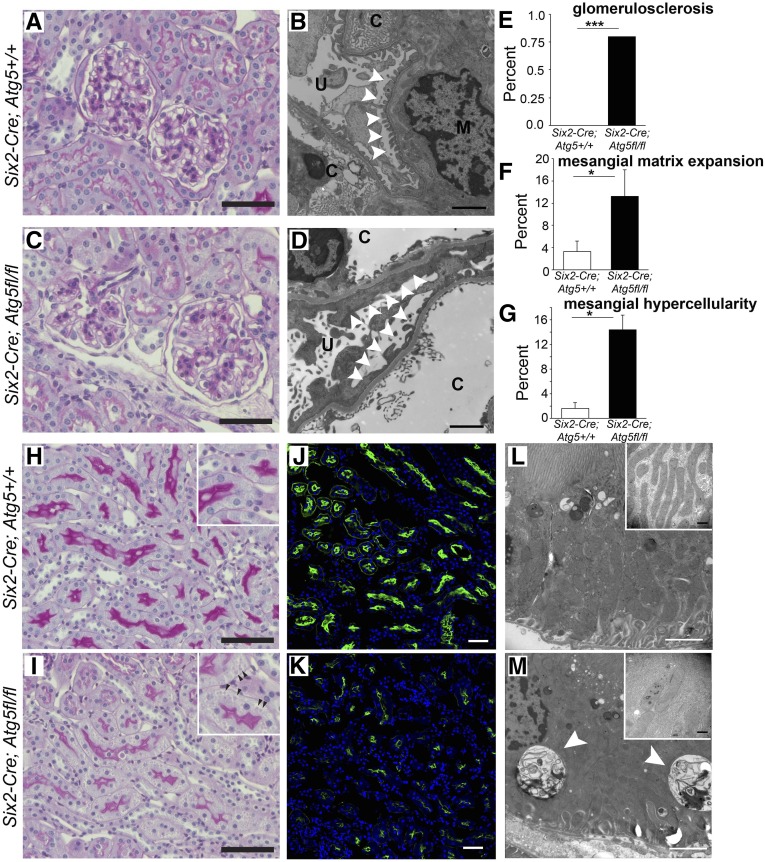
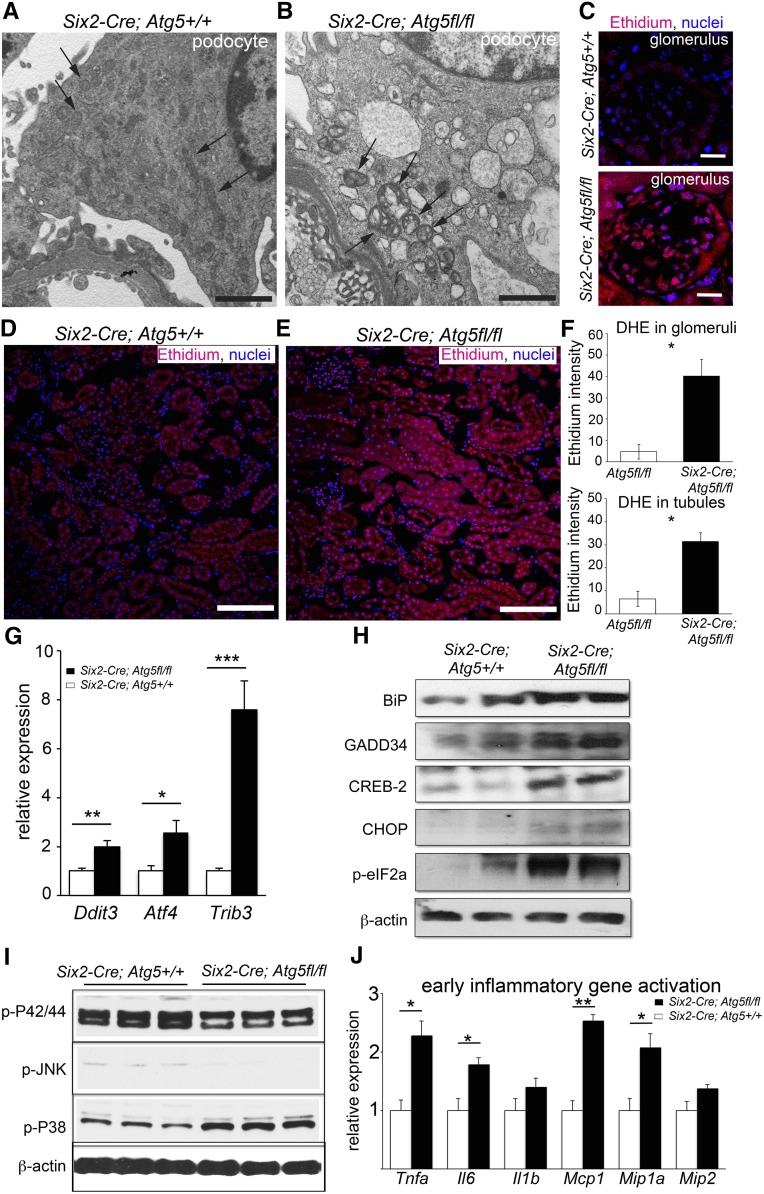
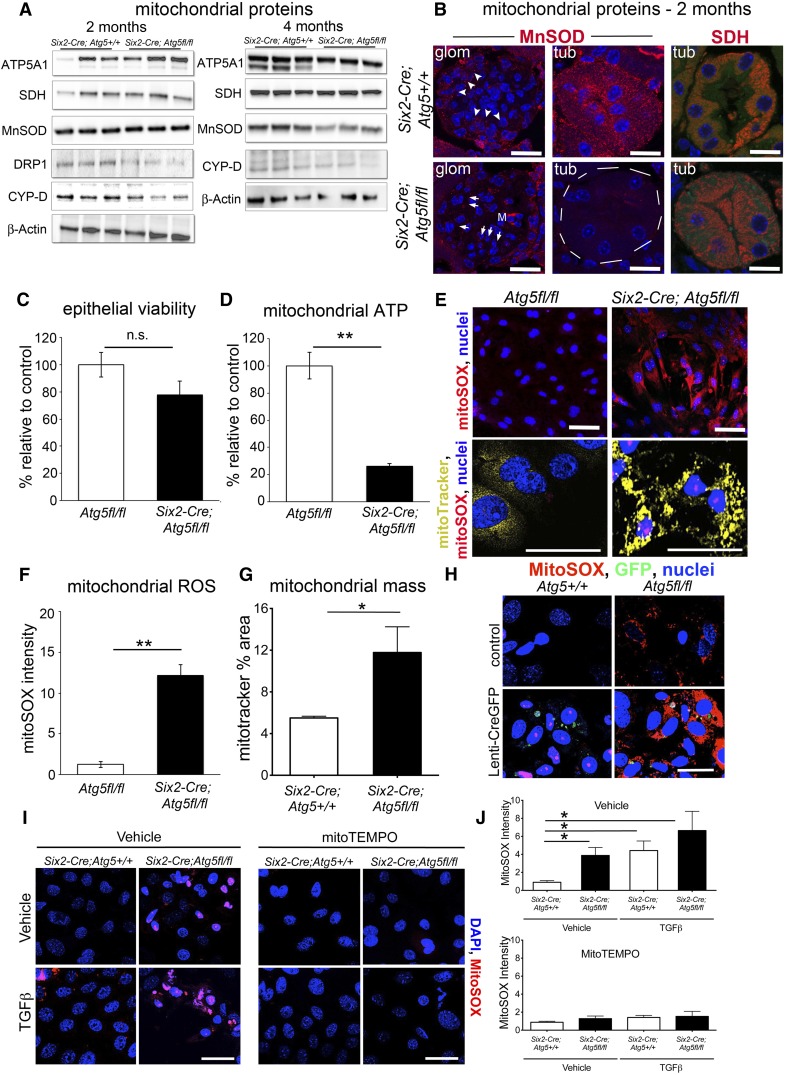
FSGS is a heterogeneous fibrosing disease of the kidney, the cause of which remains poorly understood. In most cases, there is no effective treatment to halt or retard progression to renal failure. Increasing evidence points to mitochondrial dysfunction and the generation of reactive oxygen species in the pathogenesis of CKD. Autophagy, a major intracellular lysosomal degradation system, performs homeostatic functions linked to metabolism and organelle turnover. We prevented normal autophagic pathways in nephrons of mice by mutating critical autophagy genes ATG5 or ATG7 during nephrogenesis. Mutant mice developed mild podocyte and tubular dysfunction within 2 months, profound glomerular and tubular changes bearing close similarity to human disease by 4 months, and organ failure by 6 months. Ultrastructurally, podocytes and tubular cells showed vacuolization, abnormal mitochondria, and evidence of endoplasmic reticulum stress, features that precede the appearance of histologic or clinical disease. Similar changes were observed in human idiopathic FSGS kidney biopsy specimens. Biochemical analysis of podocytes and tubules of 2-month-old mutant mice revealed elevated production of reactive oxygen species, activation of endoplasmic reticulum stress pathways, phosphorylation of p38, and mitochondrial dysfunction. Furthermore, cultured proximal tubule cells isolated from mutant mice showed marked mitochondrial dysfunction and elevated mitochondrial reactive oxygen species generation that was suppressed by a mitochondrial superoxide scavenger. We conclude that mitochondrial dysfunction and endoplasmic reticulum stress due to impaired autophagic organelle turnover in podocytes and tubular epithelium are sufficient to cause many of the manifestations of FSGS in mice.
Keywords: Autophagy; focal segmental glomerulosclerosis; mitochondria.
Copyright © 2015 by the American Society of Nephrology.
Figures
Comment in
-
Could Autophagic Exhaustion Be a Final Common Pathway for Podocytopathy in FSGS?J Am Soc Nephrol. 2015 May;26(5):999-1001. doi: 10.1681/ASN.2014090919. Epub 2014 Nov 18. J Am Soc Nephrol. 2015. PMID: 25406340 Free PMC article. No abstract available.
-
Glomerular disease: autophagy failure and mitochondrial dysfunction in FSGS.Nat Rev Nephrol. 2015 Feb;11(2):66. doi: 10.1038/nrneph.2014.233. Epub 2014 Dec 9. Nat Rev Nephrol. 2015. PMID: 25488857 No abstract available.
References
-
- Pollak MR: The Genetic Basis of FSGS and Steroid-Resistant Nephrosis. Seminars in Nephrology, New York, Elsevier, 2003, pp 141–146 - PubMed
-
- Genovese G, Friedman DJ, Ross MD, Lecordier L, Uzureau P, Freedman BI, Bowden DW, Langefeld CD, Oleksyk TK, Uscinski Knob AL, Bernhardy AJ, Hicks PJ, Nelson GW, Vanhollebeke B, Winkler CA, Kopp JB, Pays E, Pollak MR: Association of trypanolytic ApoL1 variants with kidney disease in African Americans. Science 329: 841–845, 2010 - PMC - PubMed
-
- McVicar M, Exeni R, Susin M: Nephrotic syndrome and multiple tubular defects in children: an early sign of focal segmental glomerulosclerosis. J Pediatr 97: 918–922, 1980 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
