

BAsE-Seq: a method for obtaining long viral haplotypes from short sequence reads
- PMID: 25406369
- PMCID: PMC4269956
- DOI: 10.1186/PREACCEPT-6768001251451949
BAsE-Seq: a method for obtaining long viral haplotypes from short sequence reads
Abstract
We present a method for obtaining long haplotypes, of over 3 kb in length, using a short-read sequencer, Barcode-directed Assembly for Extra-long Sequences (BAsE-Seq). BAsE-Seq relies on transposing a template-specific barcode onto random segments of the template molecule and assembling the barcoded short reads into complete haplotypes. We applied BAsE-Seq on mixed clones of hepatitis B virus and accurately identified haplotypes occurring at frequencies greater than or equal to 0.4%, with >99.9% specificity. Applying BAsE-Seq to a clinical sample, we obtained over 9,000 viral haplotypes, which provided an unprecedented view of hepatitis B virus population structure during chronic infection. BAsE-Seq is readily applicable for monitoring quasispecies evolution in viral diseases.
Trial registration: ClinicalTrials.gov NCT00962871.
Figures
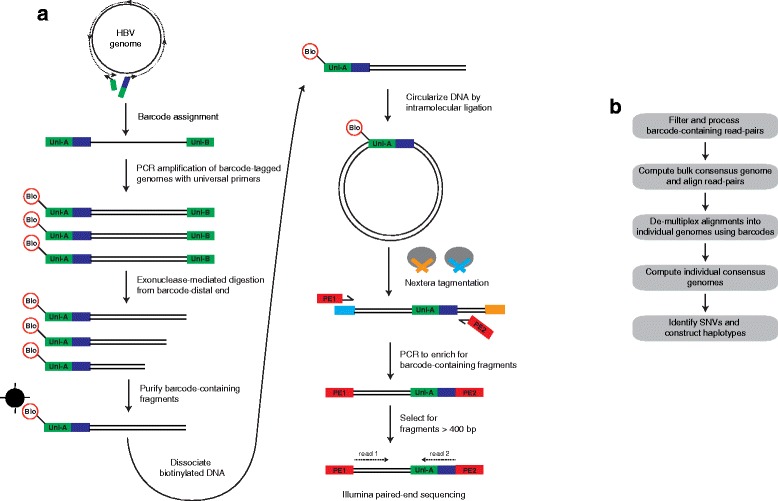
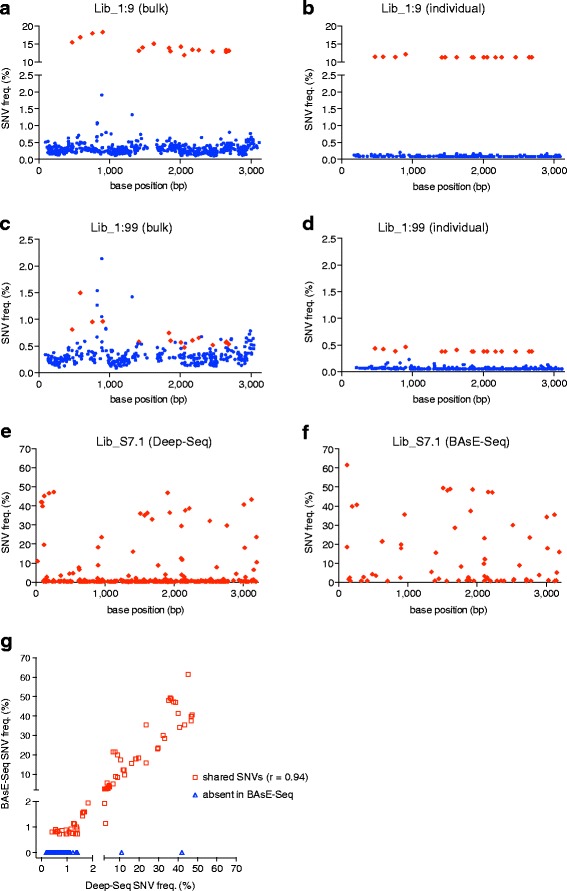

Similar articles
-
Early MinION™ nanopore single-molecule sequencing technology enables the characterization of hepatitis B virus genetic complexity in clinical samples.PLoS One. 2018 Mar 22;13(3):e0194366. doi: 10.1371/journal.pone.0194366. eCollection 2018. PLoS One. 2018. PMID: 29566006 Free PMC article.
-
Quantifying perinatal transmission of Hepatitis B viral quasispecies by tag linkage deep sequencing.Sci Rep. 2017 Aug 31;7(1):10168. doi: 10.1038/s41598-017-10591-9. Sci Rep. 2017. PMID: 28860476 Free PMC article.
-
Reconstructing viral quasispecies from NGS amplicon reads.In Silico Biol. 2011-2012;11(5-6):237-49. doi: 10.3233/ISB-2012-0458. In Silico Biol. 2011. PMID: 23202425
-
TruSPAdes: barcode assembly of TruSeq synthetic long reads.Nat Methods. 2016 Mar;13(3):248-50. doi: 10.1038/nmeth.3737. Epub 2016 Feb 1. Nat Methods. 2016. PMID: 26828418
-
Alignment of Next-Generation Sequencing Reads.Annu Rev Genomics Hum Genet. 2015;16:133-51. doi: 10.1146/annurev-genom-090413-025358. Epub 2015 May 4. Annu Rev Genomics Hum Genet. 2015. PMID: 25939052 Review.
Cited by
-
Enhancing the accuracy of next-generation sequencing for detecting rare and subclonal mutations.Nat Rev Genet. 2018 May;19(5):269-285. doi: 10.1038/nrg.2017.117. Epub 2018 Mar 26. Nat Rev Genet. 2018. PMID: 29576615 Free PMC article. Review.
-
IFDlong: an isoform and fusion detector for accurate annotation and quantification of long-read RNA-seq data.bioRxiv [Preprint]. 2024 May 14:2024.05.11.593690. doi: 10.1101/2024.05.11.593690. bioRxiv. 2024. PMID: 38798496 Free PMC article. Preprint.
-
Genomic approaches for understanding dengue: insights from the virus, vector, and host.Genome Biol. 2016 Mar 2;17:38. doi: 10.1186/s13059-016-0907-2. Genome Biol. 2016. PMID: 26931545 Free PMC article. Review.
-
Targeted transcriptome analysis using synthetic long read sequencing uncovers isoform reprograming in the progression of colon cancer.Commun Biol. 2021 Apr 27;4(1):506. doi: 10.1038/s42003-021-02024-1. Commun Biol. 2021. PMID: 33907296 Free PMC article.
-
MrHAMER yields highly accurate single molecule viral sequences enabling analysis of intra-host evolution.Nucleic Acids Res. 2021 Jul 9;49(12):e70. doi: 10.1093/nar/gkab231. Nucleic Acids Res. 2021. PMID: 33849057 Free PMC article.
References
Publication types
MeSH terms
Associated data
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
