

Rapid evaluation and quality control of next generation sequencing data with FaQCs
- PMID: 25408143
- PMCID: PMC4246454
- DOI: 10.1186/s12859-014-0366-2
Rapid evaluation and quality control of next generation sequencing data with FaQCs
Abstract
Background: Next generation sequencing (NGS) technologies that parallelize the sequencing process and produce thousands to millions, or even hundreds of millions of sequences in a single sequencing run, have revolutionized genomic and genetic research. Because of the vagaries of any platform's sequencing chemistry, the experimental processing, machine failure, and so on, the quality of sequencing reads is never perfect, and often declines as the read is extended. These errors invariably affect downstream analysis/application and should therefore be identified early on to mitigate any unforeseen effects.
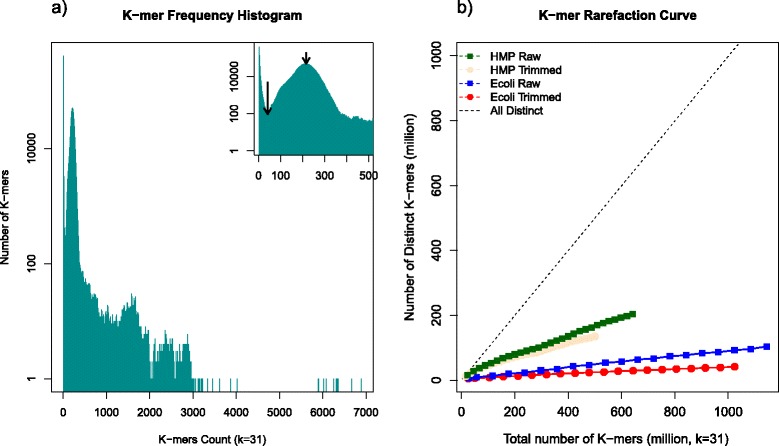
Results: Here we present a novel FastQ Quality Control Software (FaQCs) that can rapidly process large volumes of data, and which improves upon previous solutions to monitor the quality and remove poor quality data from sequencing runs. Both the speed of processing and the memory footprint of storing all required information have been optimized via algorithmic and parallel processing solutions. The trimmed output compared side-by-side with the original data is part of the automated PDF output. We show how this tool can help data analysis by providing a few examples, including an increased percentage of reads recruited to references, improved single nucleotide polymorphism identification as well as de novo sequence assembly metrics.
Conclusion: FaQCs combines several features of currently available applications into a single, user-friendly process, and includes additional unique capabilities such as filtering the PhiX control sequences, conversion of FASTQ formats, and multi-threading. The original data and trimmed summaries are reported within a variety of graphics and reports, providing a simple way to do data quality control and assurance.
Figures


References
-
- Kwon S, Park S, Lee B, Yoon S. In-depth analysis of interrelation between quality scores and real errors in illumina reads. Conf Proc IEEE Eng Med Biol Soc. 2013;2013:635–638. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
