

Indel reliability in indel-based phylogenetic inference
- PMID: 25409663
- PMCID: PMC4986452
- DOI: 10.1093/gbe/evu252
Indel reliability in indel-based phylogenetic inference
Abstract
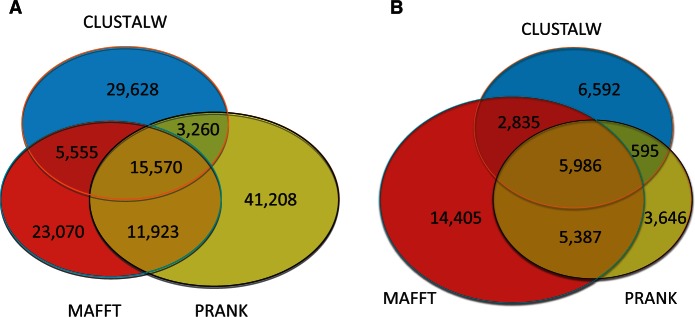
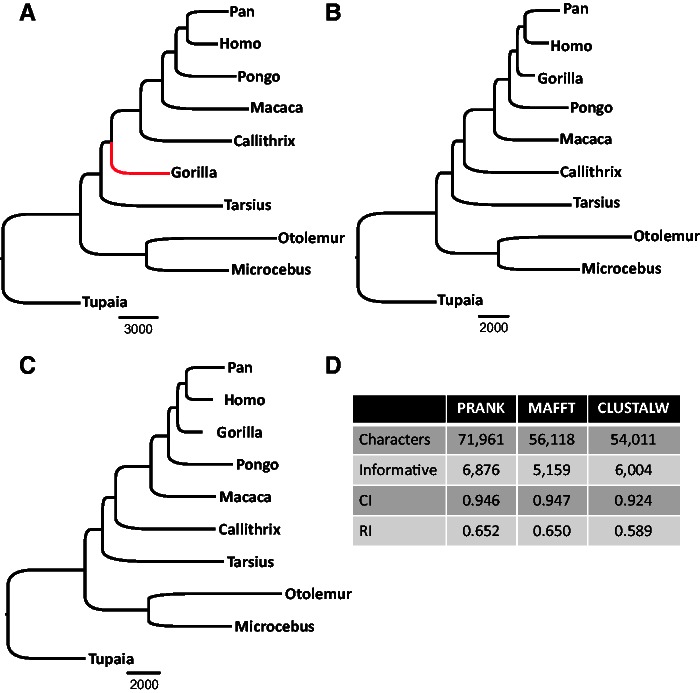
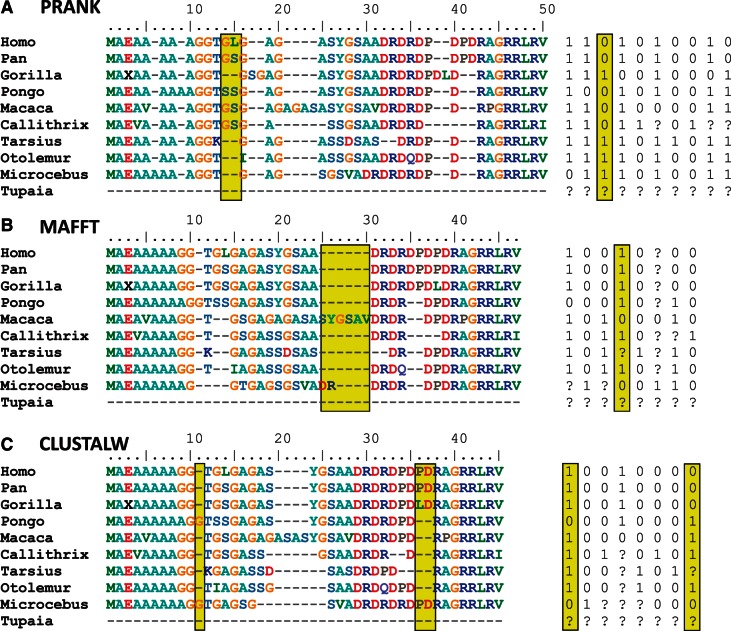
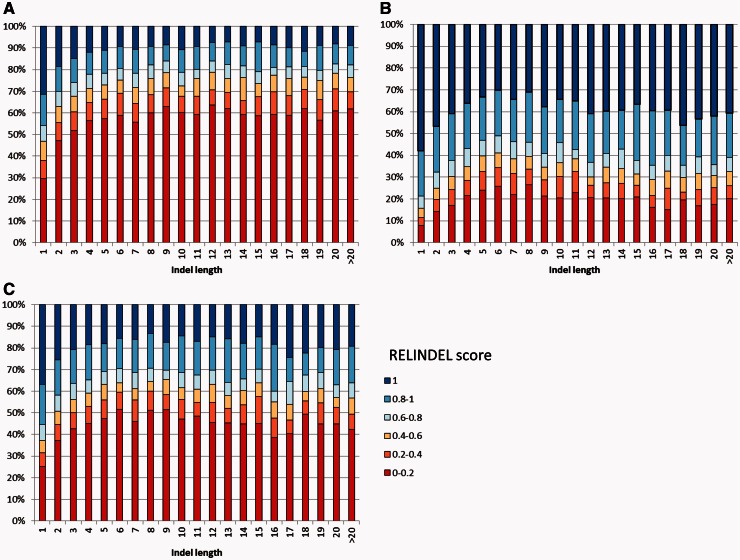
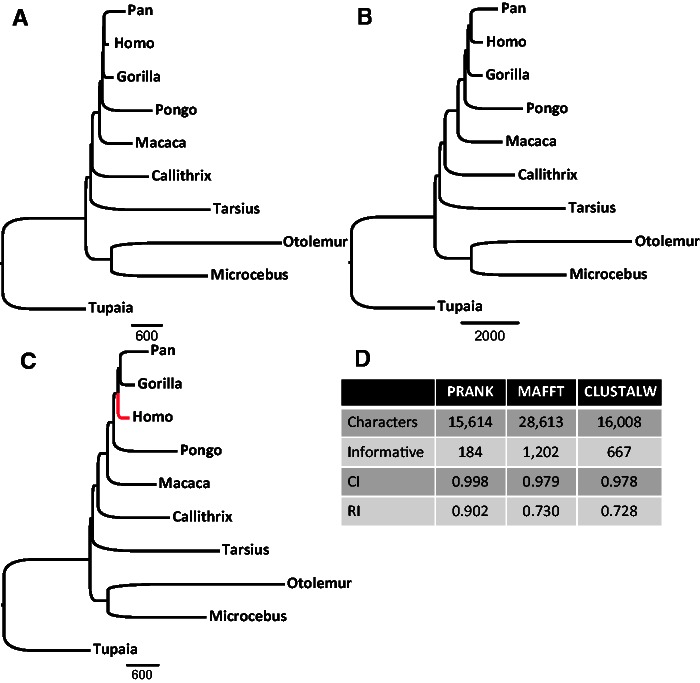
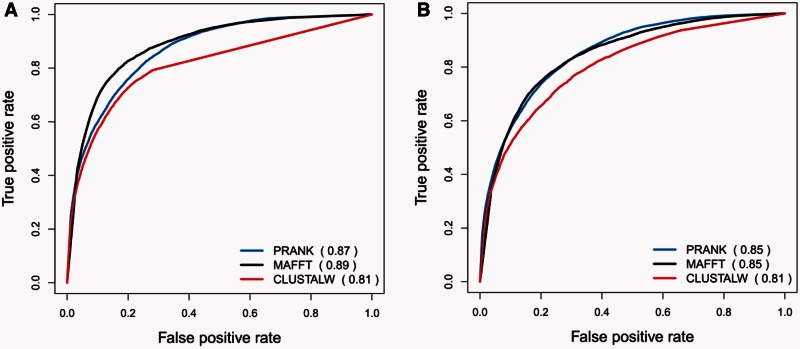
It is often assumed that it is unlikely that the same insertion or deletion (indel) event occurred at the same position in two independent evolutionary lineages, and thus, indel-based inference of phylogeny should be less subject to homoplasy compared with standard inference which is based on substitution events. Indeed, indels were successfully used to solve debated evolutionary relationships among various taxonomical groups. However, indels are never directly observed but rather inferred from the alignment and thus indel-based inference may be sensitive to alignment errors. It is hypothesized that phylogenetic reconstruction would be more accurate if it relied only on a subset of reliable indels instead of the entire indel data. Here, we developed a method to quantify the reliability of indel characters by measuring how often they appear in a set of alternative multiple sequence alignments. Our approach is based on the assumption that indels that are consistently present in most alternative alignments are more reliable compared with indels that appear only in a small subset of these alignments. Using simulated and empirical data, we studied the impact of filtering and weighting indels by their reliability scores on the accuracy of indel-based phylogenetic reconstruction. The new method is available as a web-server at http://guidance.tau.ac.il/RELINDEL/.
Keywords: alignment reliability; indel analysis; multiple sequence alignment; phylogeny.
© The Author(s) 2014. Published by Oxford University Press on behalf of the Society for Molecular Biology and Evolution.
Figures
References
-
- Bapteste E, Philippe H. The potential value of indels as phylogenetic markers: position of trichomonads as a case study. Mol Biol Evol. 2002;19:972–977. - PubMed
-
- Belinky F, Cohen O, Huchon D. Large-scale parsimony analysis of metazoan indels in protein-coding genes. Mol Biol Evol. 2010;27:441–451. - PubMed
-
- Blackburne BP, Whelan S. Class of multiple sequence alignment algorithm affects genomic analysis. Mol Biol Evol. 2013;30:642–653. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
