

Topologically associating domains are stable units of replication-timing regulation
- PMID: 25409831
- PMCID: PMC4251741
- DOI: 10.1038/nature13986
Topologically associating domains are stable units of replication-timing regulation
Abstract
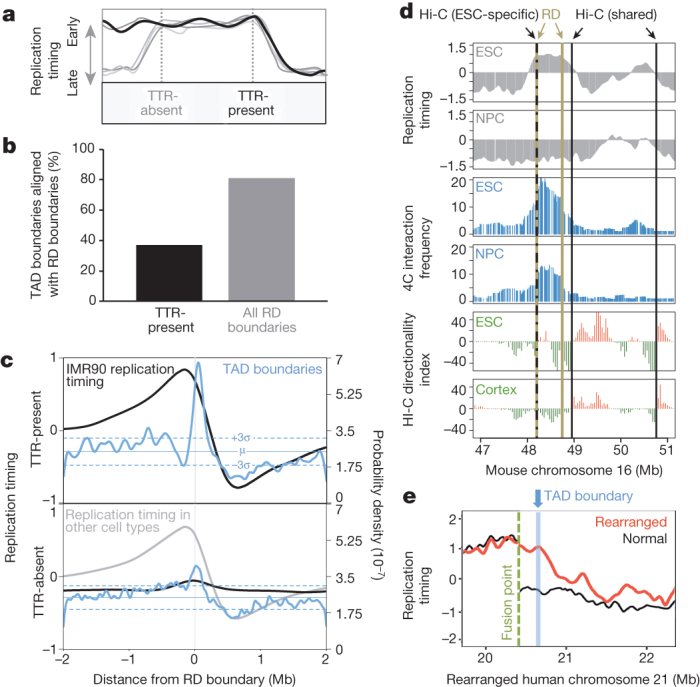
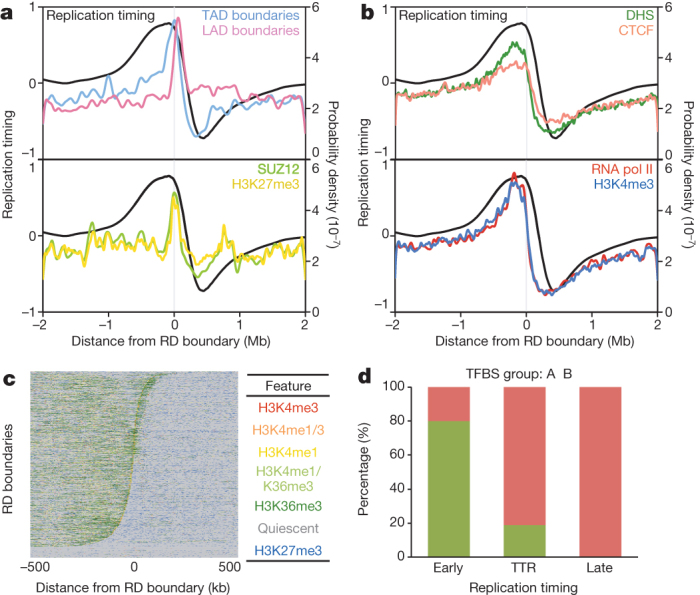
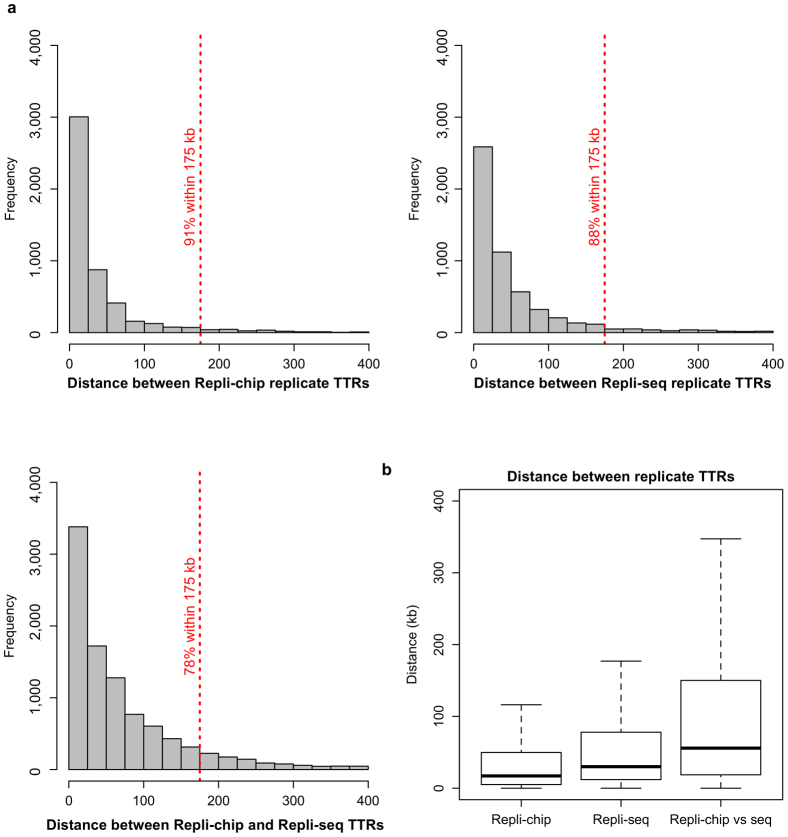
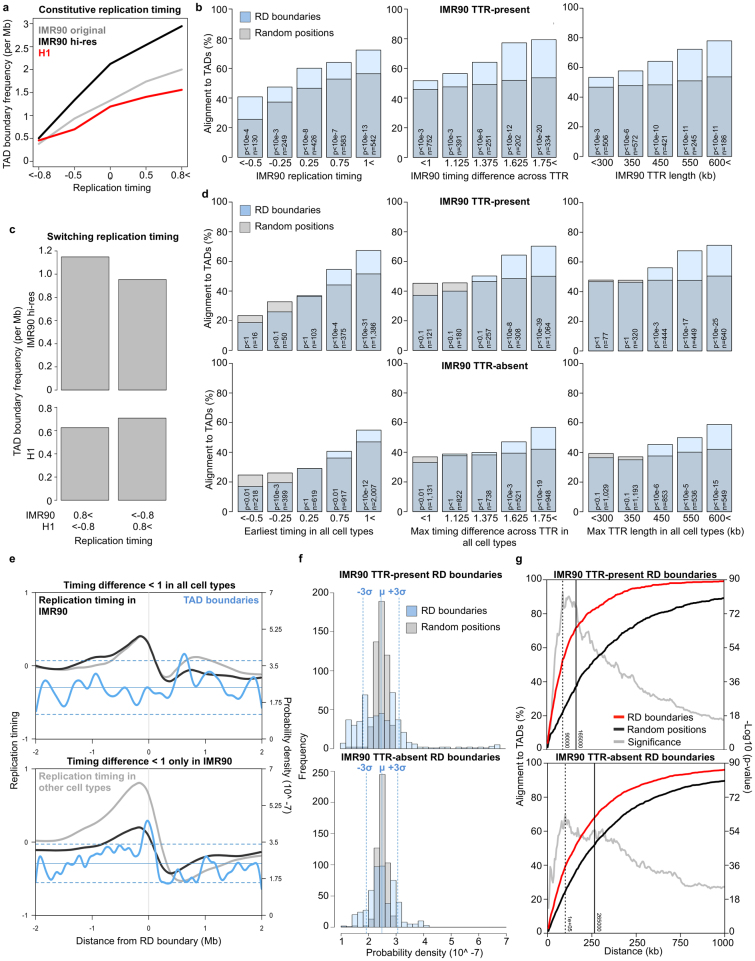
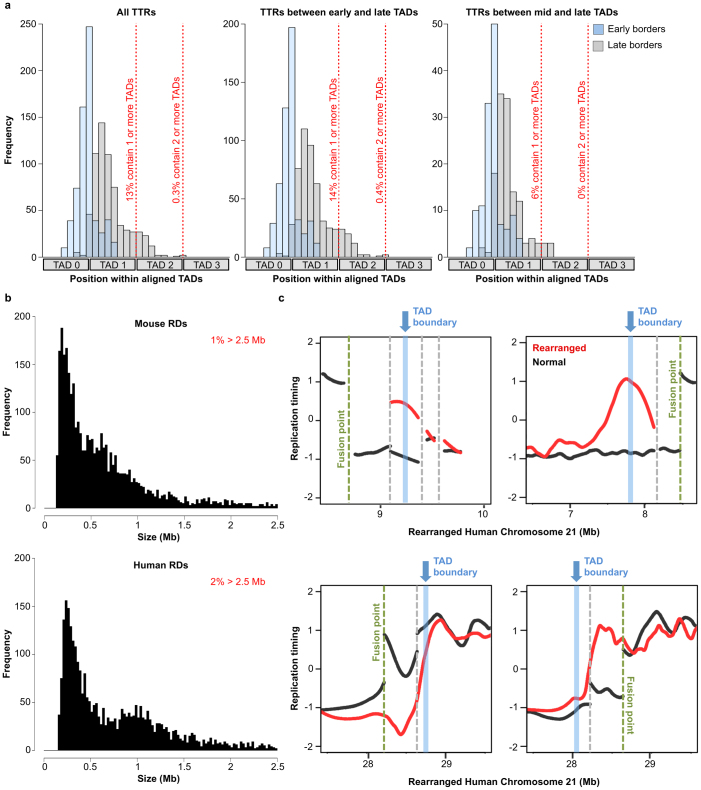
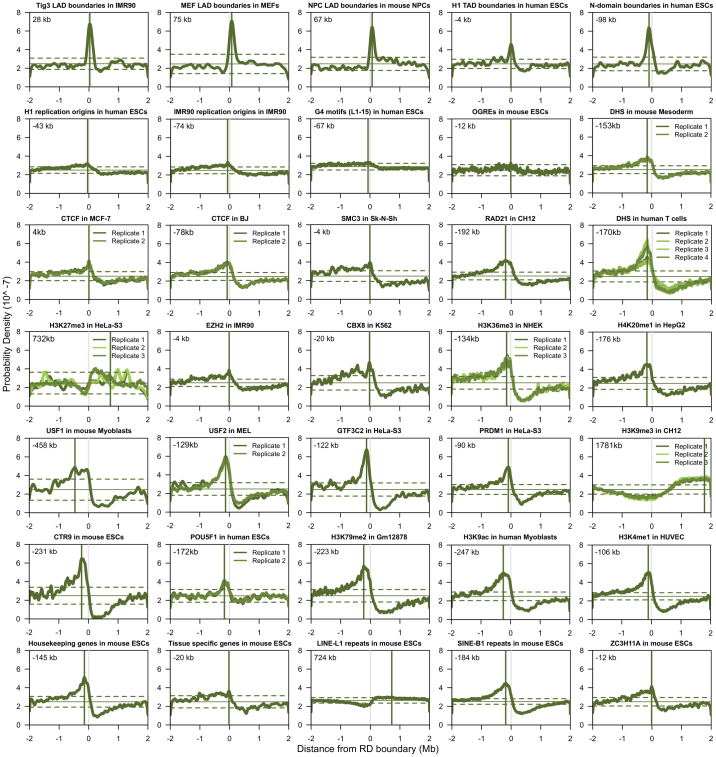
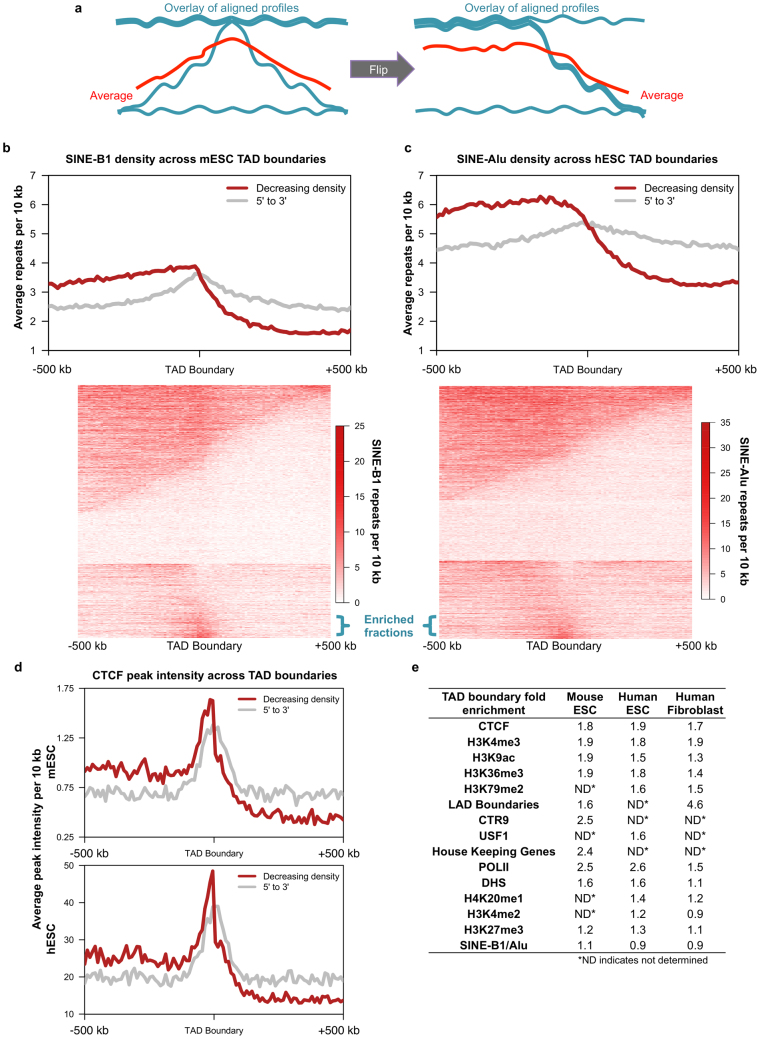
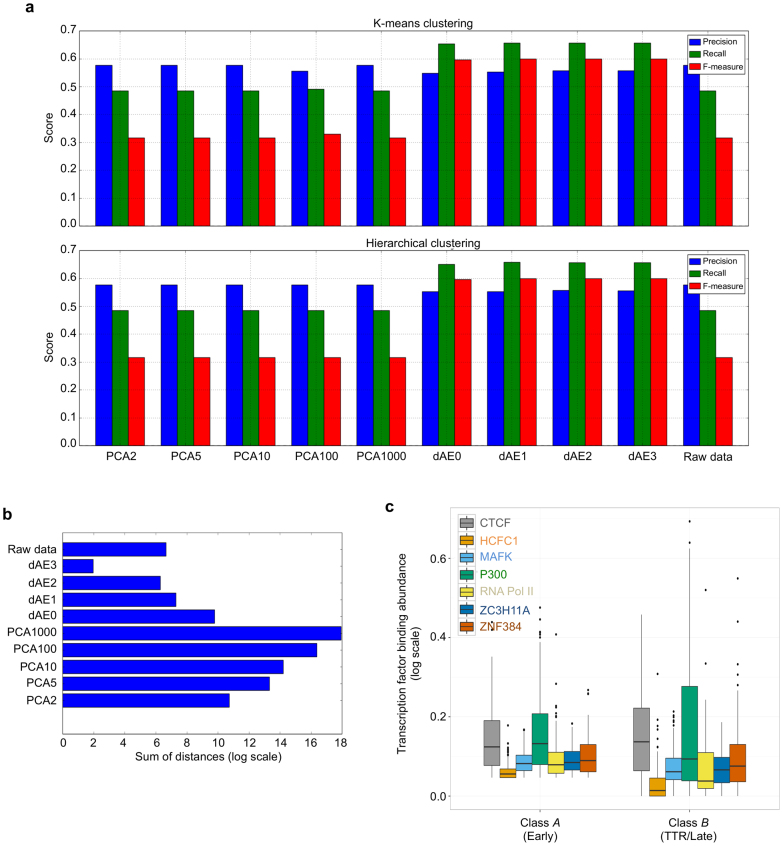
Eukaryotic chromosomes replicate in a temporal order known as the replication-timing program. In mammals, replication timing is cell-type-specific with at least half the genome switching replication timing during development, primarily in units of 400-800 kilobases ('replication domains'), whose positions are preserved in different cell types, conserved between species, and appear to confine long-range effects of chromosome rearrangements. Early and late replication correlate, respectively, with open and closed three-dimensional chromatin compartments identified by high-resolution chromosome conformation capture (Hi-C), and, to a lesser extent, late replication correlates with lamina-associated domains (LADs). Recent Hi-C mapping has unveiled substructure within chromatin compartments called topologically associating domains (TADs) that are largely conserved in their positions between cell types and are similar in size to replication domains. However, TADs can be further sub-stratified into smaller domains, challenging the significance of structures at any particular scale. Moreover, attempts to reconcile TADs and LADs to replication-timing data have not revealed a common, underlying domain structure. Here we localize boundaries of replication domains to the early-replicating border of replication-timing transitions and map their positions in 18 human and 13 mouse cell types. We demonstrate that, collectively, replication domain boundaries share a near one-to-one correlation with TAD boundaries, whereas within a cell type, adjacent TADs that replicate at similar times obscure replication domain boundaries, largely accounting for the previously reported lack of alignment. Moreover, cell-type-specific replication timing of TADs partitions the genome into two large-scale sub-nuclear compartments revealing that replication-timing transitions are indistinguishable from late-replicating regions in chromatin composition and lamina association and accounting for the reduced correlation of replication timing to LADs and heterochromatin. Our results reconcile cell-type-specific sub-nuclear compartmentalization and replication timing with developmentally stable structural domains and offer a unified model for large-scale chromosome structure and function.
Conflict of interest statement
The authors declare no competing financial interests.
Figures





References
-
- Wright ML, Grützner F, Seligmann H. DNA Replication Current Advances. 2011.
Publication types
MeSH terms
Substances
Grants and funding
- HG003991/HG/NHGRI NIH HHS/United States
- F31 CA165863/CA/NCI NIH HHS/United States
- DK065806/DK/NIDDK NIH HHS/United States
- RC2 HG005573/HG/NHGRI NIH HHS/United States
- R56 DK065806/DK/NIDDK NIH HHS/United States
- HG005573/HG/NHGRI NIH HHS/United States
- F31CA165863/CA/NCI NIH HHS/United States
- P01 GM085354/GM/NIGMS NIH HHS/United States
- R01 DA033775/DA/NIDA NIH HHS/United States
- GM085354/GM/NIGMS NIH HHS/United States
- HG005602/HG/NHGRI NIH HHS/United States
- R01 DK065806/DK/NIDDK NIH HHS/United States
- U54 HG006997/HG/NHGRI NIH HHS/United States
- R01 GM083337/GM/NIGMS NIH HHS/United States
- R01 HG003991/HG/NHGRI NIH HHS/United States
- GM083337/GM/NIGMS NIH HHS/United States
- RC2 HG005602/HG/NHGRI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
