

Dynamic analyses of alternative polyadenylation from RNA-seq reveal a 3'-UTR landscape across seven tumour types
- PMID: 25409906
- PMCID: PMC4467577
- DOI: 10.1038/ncomms6274
Dynamic analyses of alternative polyadenylation from RNA-seq reveal a 3'-UTR landscape across seven tumour types
Abstract
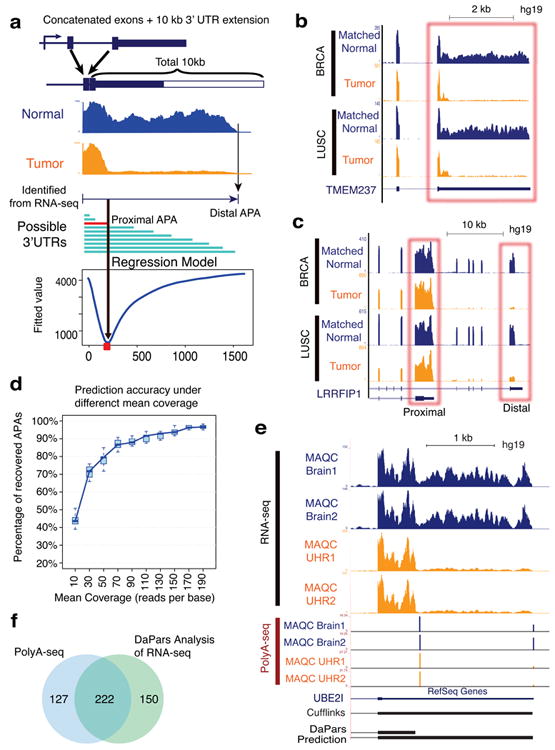
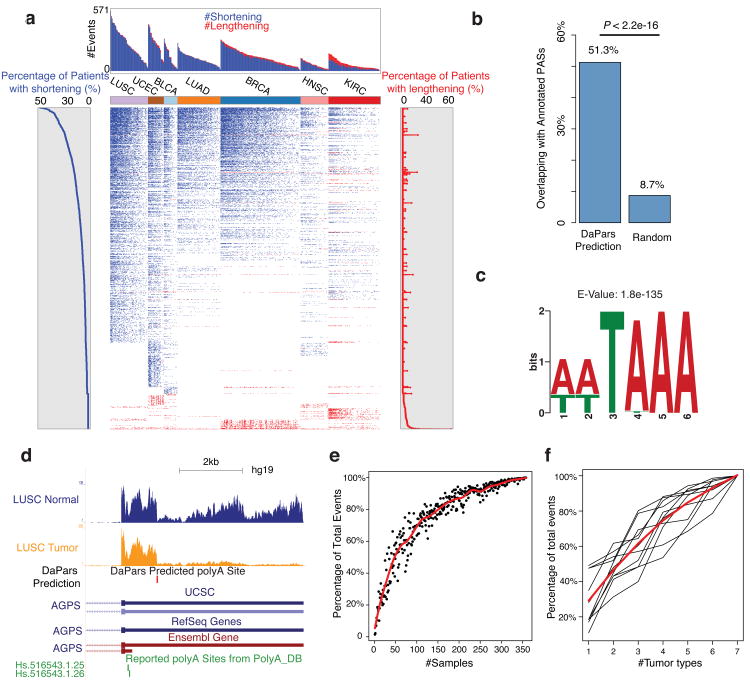
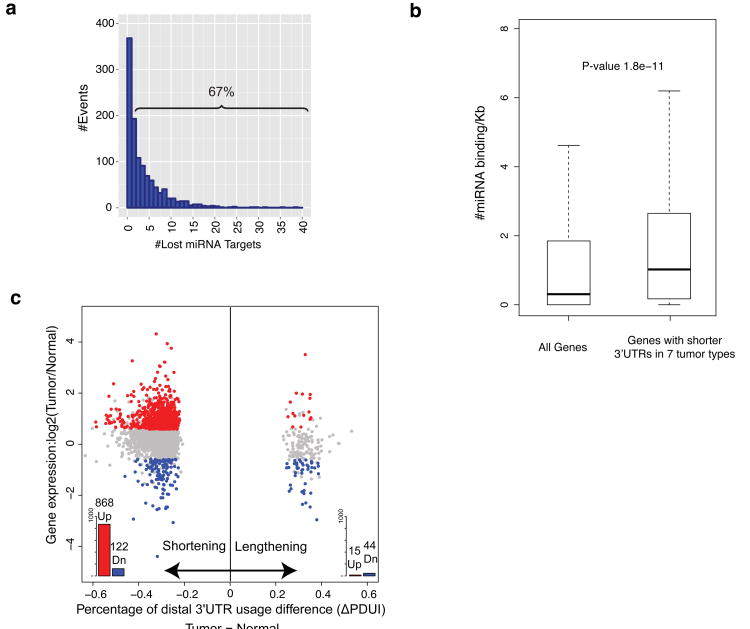
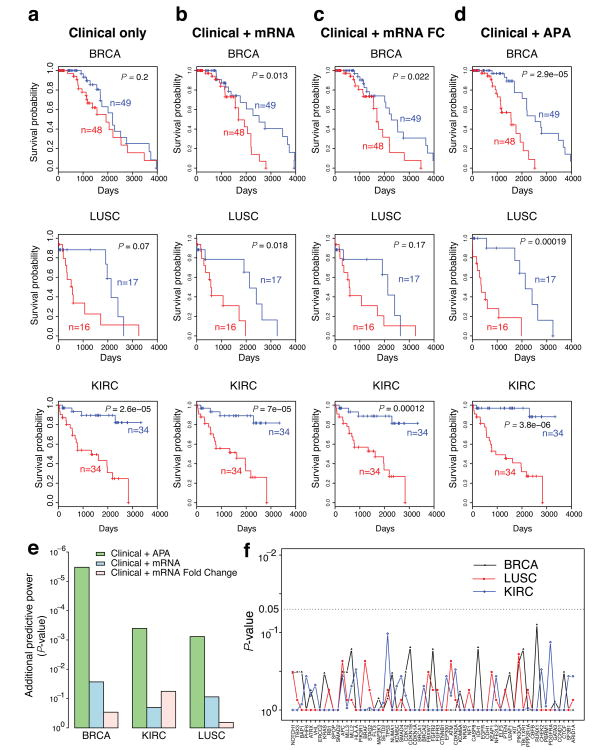
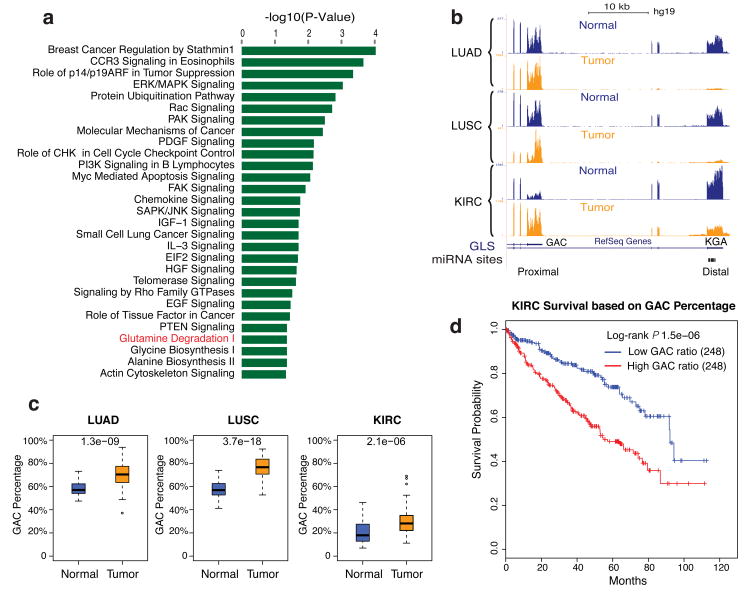
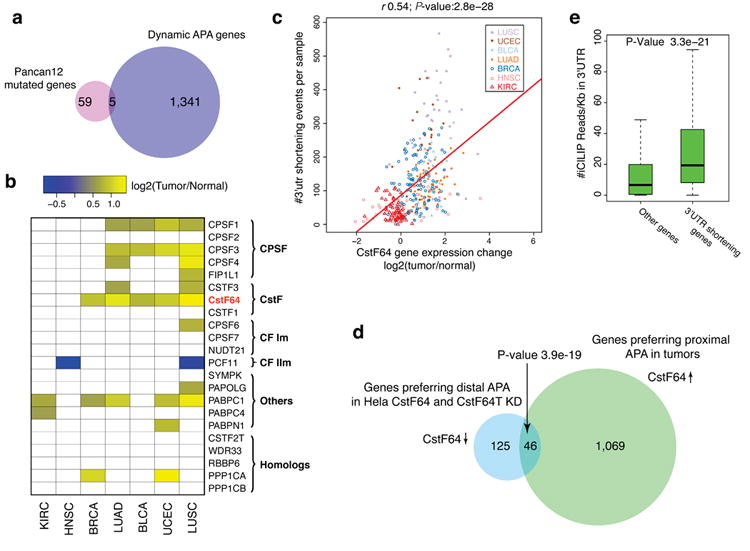
Alternative polyadenylation (APA) is a pervasive mechanism in the regulation of most human genes, and its implication in diseases including cancer is only beginning to be appreciated. Since conventional APA profiling has not been widely adopted, global cancer APA studies are very limited. Here we develop a novel bioinformatics algorithm (DaPars) for the de novo identification of dynamic APAs from standard RNA-seq. When applied to 358 TCGA Pan-Cancer tumour/normal pairs across seven tumour types, DaPars reveals 1,346 genes with recurrent and tumour-specific APAs. Most APA genes (91%) have shorter 3'-untranslated regions (3' UTRs) in tumours that can avoid microRNA-mediated repression, including glutaminase (GLS), a key metabolic enzyme for tumour proliferation. Interestingly, selected APA events add strong prognostic power beyond common clinical and molecular variables, suggesting their potential as novel prognostic biomarkers. Finally, our results implicate CstF64, an essential polyadenylation factor, as a master regulator of 3'-UTR shortening across multiple tumour types.
Figures
References
-
- Elkon R, Ugalde AP, Agami R. Alternative cleavage and polyadenylation: extent, regulation and function. Nature reviews Genetics. 2013;14:496–506. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
