

The Harvest suite for rapid core-genome alignment and visualization of thousands of intraspecific microbial genomes
- PMID: 25410596
- PMCID: PMC4262987
- DOI: 10.1186/s13059-014-0524-x
The Harvest suite for rapid core-genome alignment and visualization of thousands of intraspecific microbial genomes
Abstract
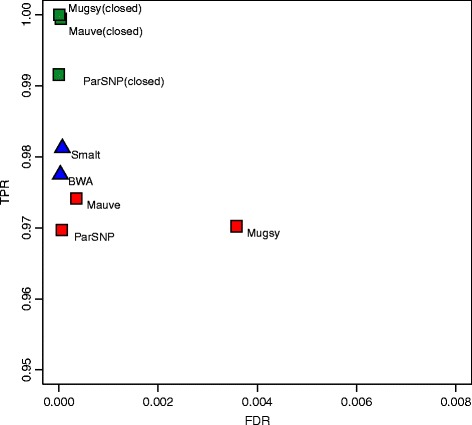
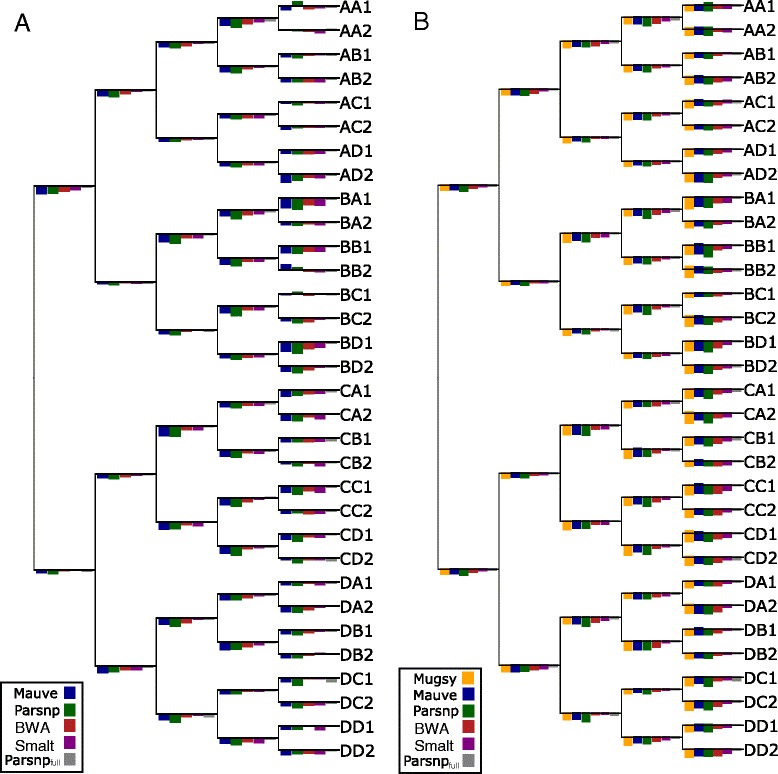
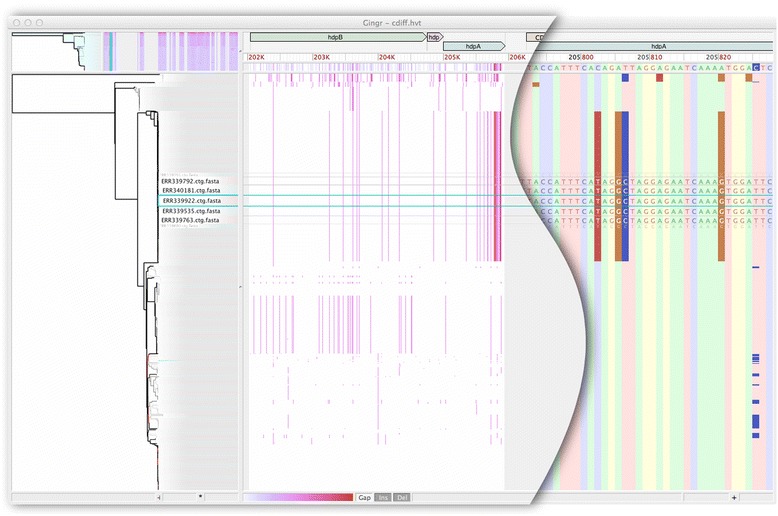
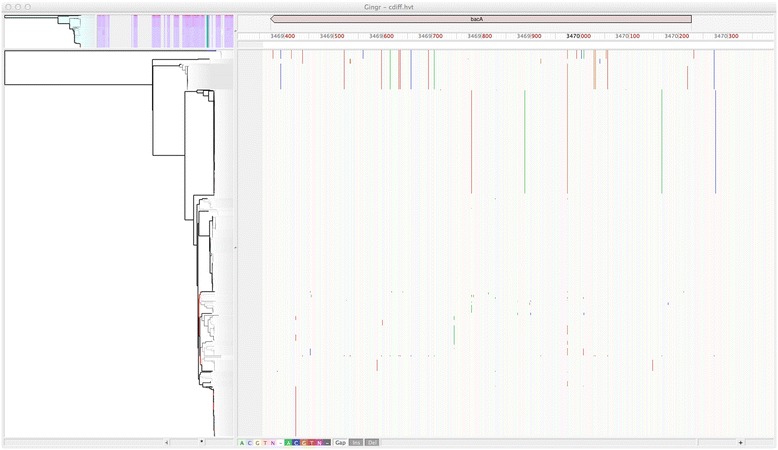
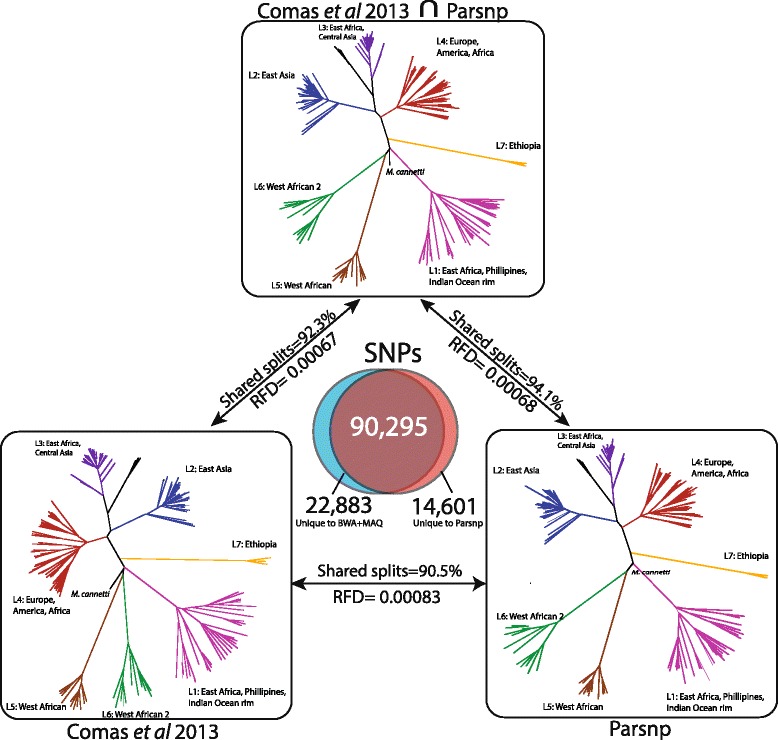
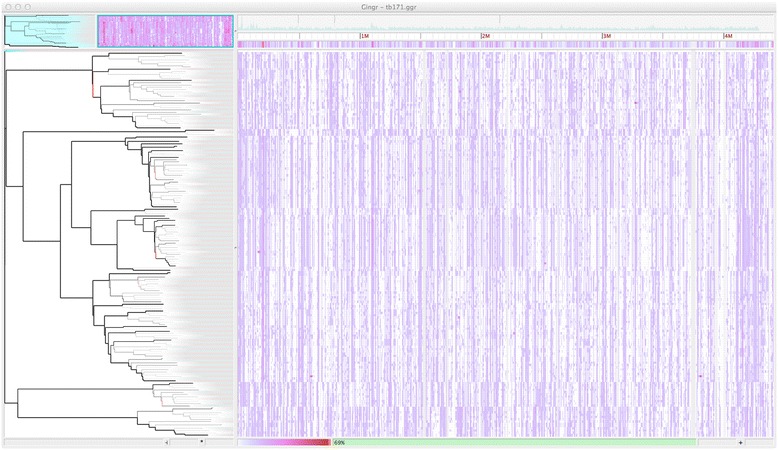
Whole-genome sequences are now available for many microbial species and clades, however existing whole-genome alignment methods are limited in their ability to perform sequence comparisons of multiple sequences simultaneously. Here we present the Harvest suite of core-genome alignment and visualization tools for the rapid and simultaneous analysis of thousands of intraspecific microbial strains. Harvest includes Parsnp, a fast core-genome multi-aligner, and Gingr, a dynamic visual platform. Together they provide interactive core-genome alignments, variant calls, recombination detection, and phylogenetic trees. Using simulated and real data we demonstrate that our approach exhibits unrivaled speed while maintaining the accuracy of existing methods. The Harvest suite is open-source and freely available from: http://github.com/marbl/harvest.
Figures
References
-
- Rasko DA, Webster DR, Sahl JW, Bashir A, Boisen N, Scheutz F, Paxinos EE, Sebra R, Chin CS, Iliopoulos D, Klammer A, Peluso P, Lee L, Kislyuk AO, Bullard J, Kasarskis A, Wang S, Eid J, Rank D, Redman JC, Steyert SR, Frimodt-Moller J, Struve C, Petersen AM, Krogfelt KA, Nataro JP, Schadt EE, Waldor MK. Origins of the E. coli strain causing an outbreak of hemolytic-uremic syndrome in Germany. N Engl J Med. 2011;365:709–717. doi: 10.1056/NEJMoa1106920. - DOI - PMC - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
