

Delayed disease onset and extended survival in the SOD1G93A rat model of amyotrophic lateral sclerosis after suppression of mutant SOD1 in the motor cortex
- PMID: 25411487
- PMCID: PMC4298650
- DOI: 10.1523/JNEUROSCI.2037-14.2014
Delayed disease onset and extended survival in the SOD1G93A rat model of amyotrophic lateral sclerosis after suppression of mutant SOD1 in the motor cortex
Abstract
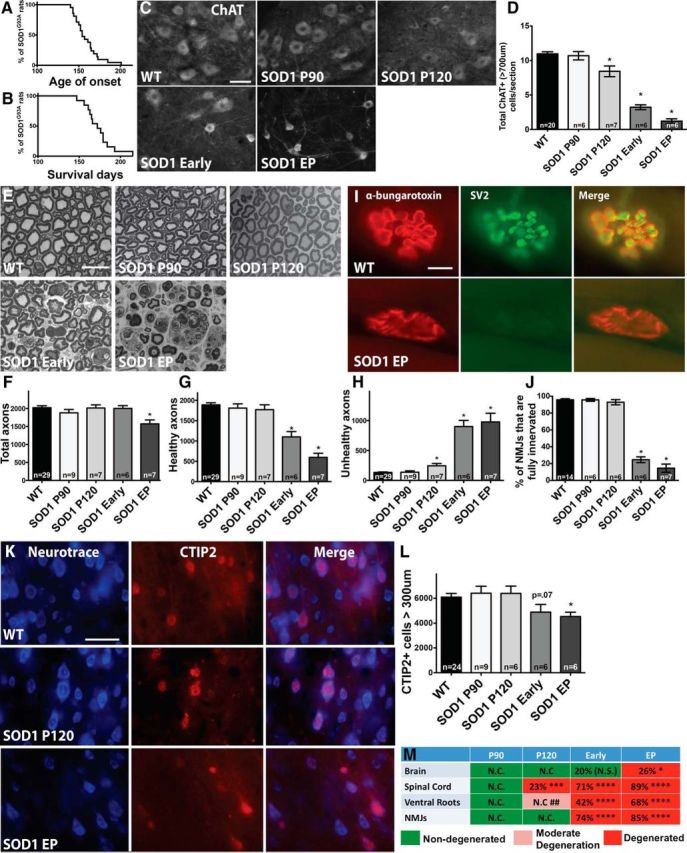
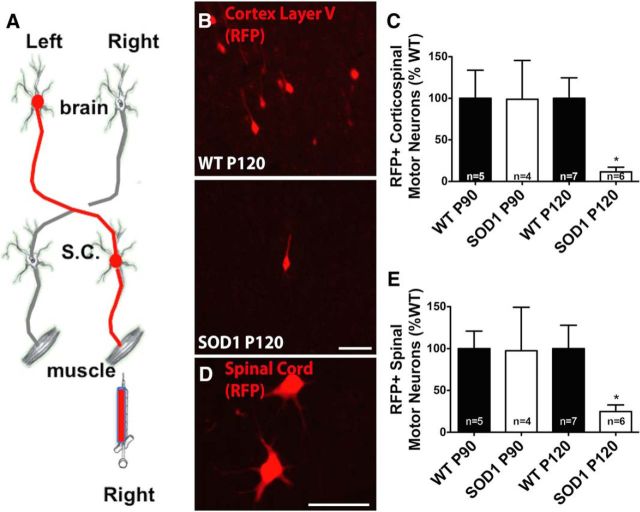
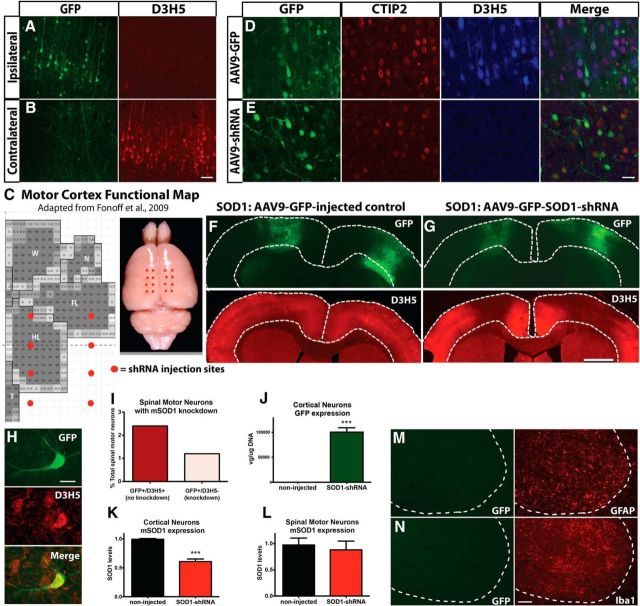
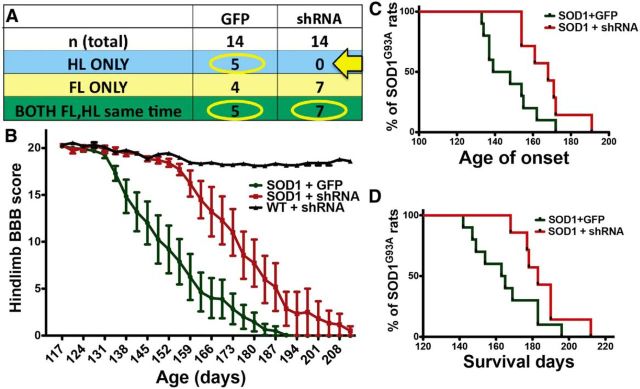
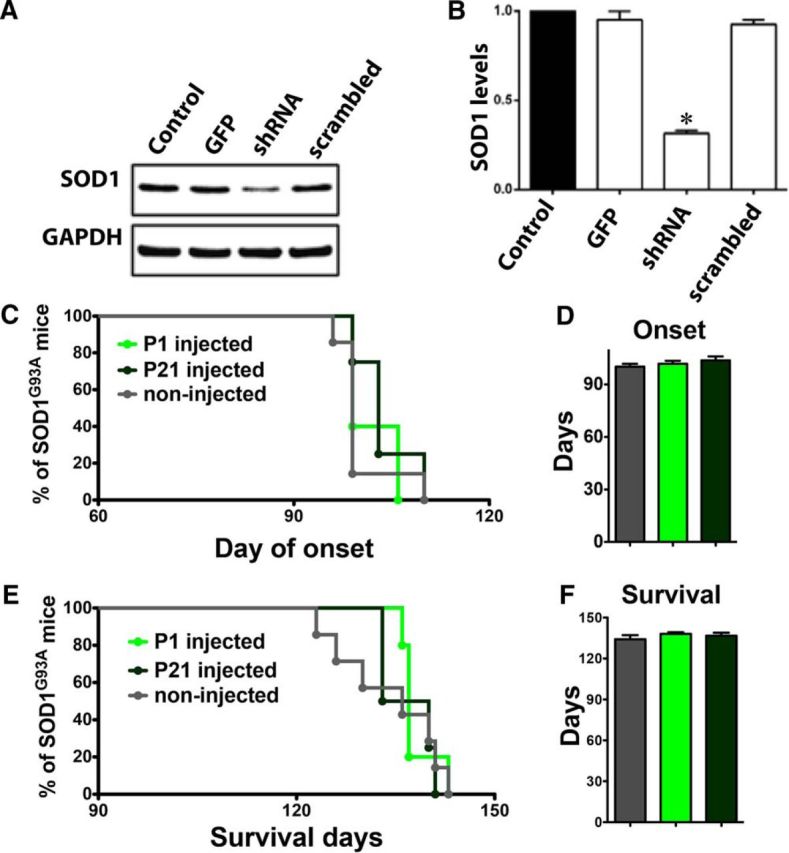
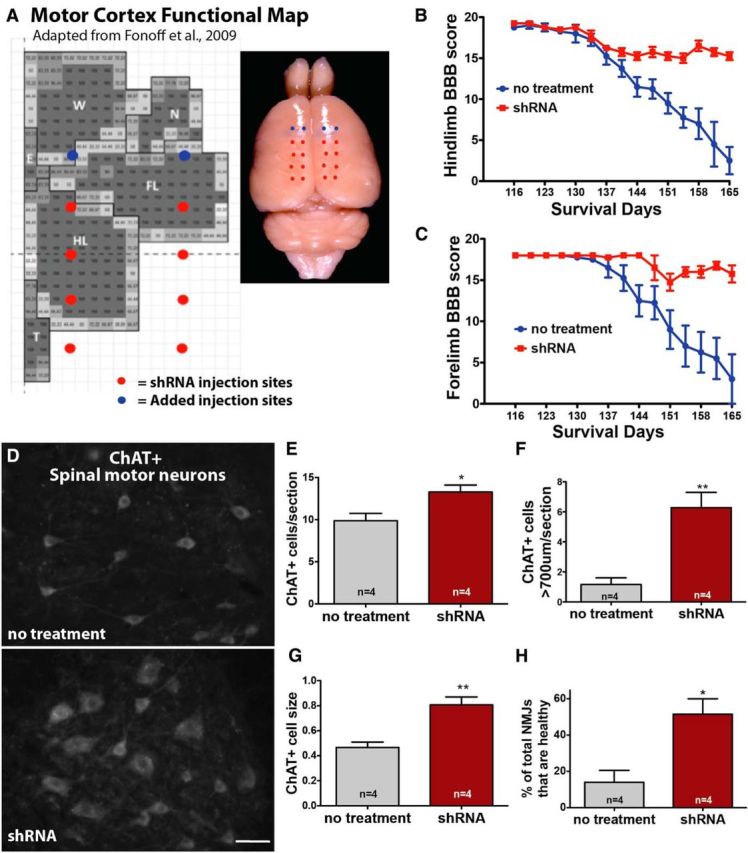
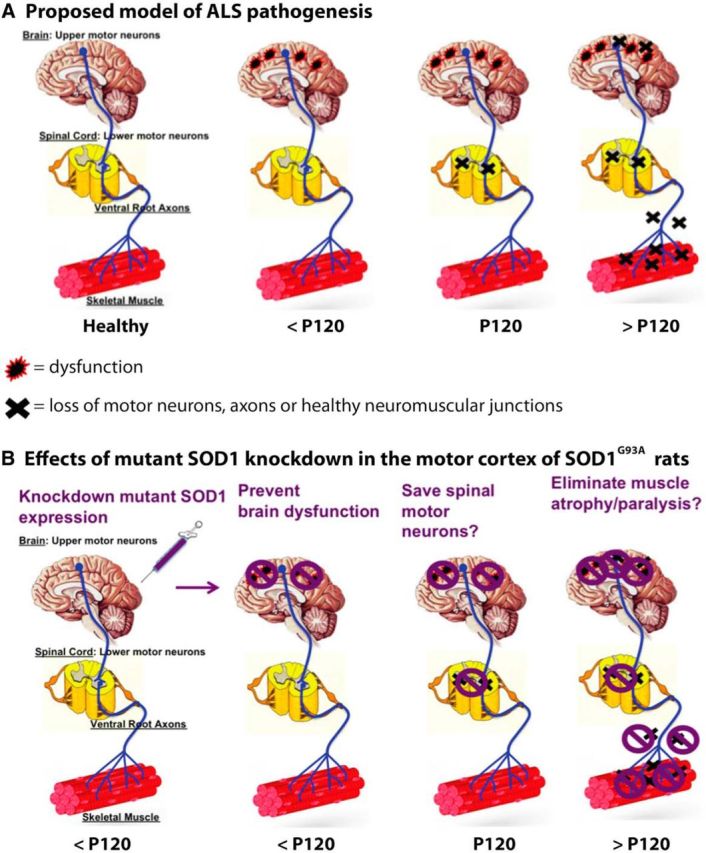
Sporadic amyotrophic lateral sclerosis (ALS) is a fatal disease with unknown etiology, characterized by a progressive loss of motor neurons leading to paralysis and death typically within 3-5 years of onset. Recently, there has been remarkable progress in understanding inherited forms of ALS in which well defined mutations are known to cause the disease. Rodent models in which the superoxide dismutase-1 (SOD1) mutation is overexpressed recapitulate hallmark signs of ALS in patients. Early anatomical changes in mouse models of fALS are seen in the neuromuscular junctions (NMJs) and lower motor neurons, and selective reduction of toxic mutant SOD1 in the spinal cord and muscle of these models has beneficial effects. Therefore, much of ALS research has focused on spinal motor neuron and NMJ aspects of the disease. Here we show that, in the SOD1(G93A) rat model of ALS, spinal motor neuron loss occurs presymptomatically and before degeneration of ventral root axons and denervation of NMJs. Although overt cell death of corticospinal motor neurons does not occur until disease endpoint, we wanted to establish whether the upper motor neuron might still play a critical role in disease progression. Surprisingly, the knockdown of mutant SOD1 in only the motor cortex of presymptomatic SOD1(G93A) rats through targeted delivery of AAV9-SOD1-shRNA resulted in a significant delay of disease onset, expansion of lifespan, enhanced survival of spinal motor neurons, and maintenance of NMJs. This datum suggests an early dysfunction and thus an important role of the upper motor neuron in this animal model of ALS and perhaps patients with the disease.
Keywords: ALS; RNAi; SOD1; amyotrophic lateral sclerosis; motor neuron disease; neurodegenerative disorder.
Copyright © 2014 the authors 0270-6474/14/3415587-14$15.00/0.
Figures
References
-
- Brockington A, Heath PR, Holden H, Kasher P, Bender FL, Claes F, Lambrechts D, Sendtner M, Carmeliet P, Shaw PJ. Downregulation of genes with a function in axon outgrowth and synapse formation in motor neurones of the VEGFdelta/delta mouse model of amyotrophic lateral sclerosis. BMC Genomics. 2010;11:203. doi: 10.1186/1471-2164-11-203. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous