

Cancer genomic research at the crossroads: realizing the changing genetic landscape as intratumoral spatial and temporal heterogeneity becomes a confounding factor
- PMID: 25411563
- PMCID: PMC4236490
- DOI: 10.1186/s12935-014-0115-7
Cancer genomic research at the crossroads: realizing the changing genetic landscape as intratumoral spatial and temporal heterogeneity becomes a confounding factor
Abstract
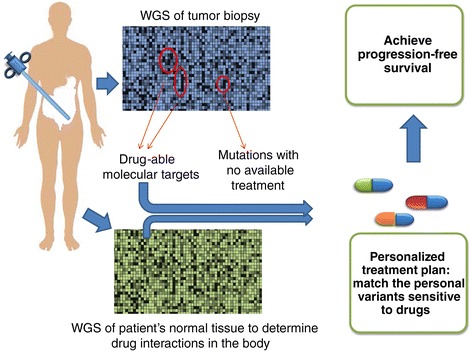
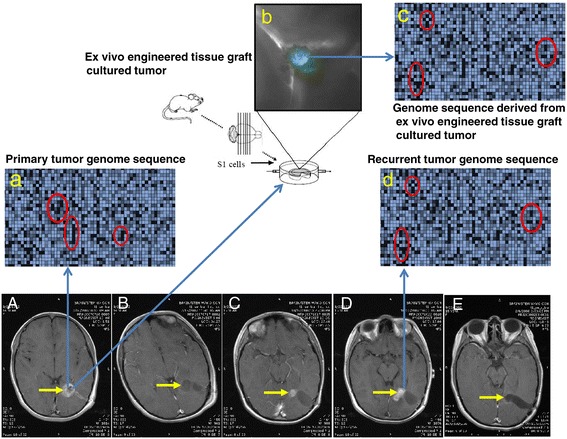
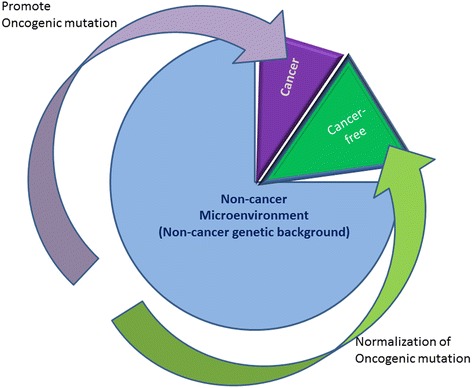
The US National Cancer Institute (NCI) and the National Human Genome Research Institute (NHGRI) created the Cancer Genome Atlas (TCGA) Project in 2006. The TCGA's goal was to sequence the genomes of 10,000 tumors to identify common genetic changes among different types of tumors for developing genetic-based treatments. TCGA offered great potential for cancer patients, but in reality has little impact on clinical applications. Recent reports place the past TCGA approach of testing a small tumor mass at a single time-point at a crossroads. This crossroads presents us with the conundrum of whether we should sequence more tumors or obtain multiple biopsies from each individual tumor at different time points. Sequencing more tumors with the past TCGA approach of single time-point sampling can neither capture the heterogeneity between different parts of the same tumor nor catch the heterogeneity that occurs as a function of time, error rates, and random drift. Obtaining multiple biopsies from each individual tumor presents multiple logistical and financial challenges. Here, we review current literature and rethink the utility and application of the TCGA approach. We discuss that the TCGA-led catalogue may provide insights into studying the functional significance of oncogenic genes in reference to non-cancer genetic background. Different methods to enhance identifying cancer targets, such as single cell technology, real time imaging of cancer cells with a biological global positioning system, and cross-referencing big data sets, are offered as ways to address sampling discrepancies in the face of tumor heterogeneity. We predict that TCGA landmarks may prove far more useful for cancer prevention than for cancer diagnosis and treatment when considering the effect of non-cancer genes and the normal genetic background on tumor microenvironment. Cancer prevention can be better realized once we understand how therapy affects the genetic makeup of cancer over time in a clinical setting. This may help create novel therapies for gene mutations that arise during a tumor's evolution from the selection pressure of treatment.
Figures
References
-
- Gerlinger M, Rowan AJ, Horswell S, Larkin J, Endesfelder D, Gronroos E, Martinez P, Matthews N, Stewart A, Tarpey P, Varela I, Phillimore B, Begum S, McDonald NQ, Butler A, Jones D, Raine K, Latimer C, Santos CR, Nohadani M, Eklund AC, Spencer-Dene B, Clark G, Pickering L, Stamp G, Gore M, Szallasi Z, Downward J, Futreal PA, Swanton C. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med. 2012;366(10):883–892. doi: 10.1056/NEJMoa1113205. - DOI - PMC - PubMed
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
