

The Genetics of Spinal Muscular Atrophy: Progress and Challenges
- PMID: 25413156
- PMCID: PMC4404441
- DOI: 10.1007/s13311-014-0314-x
The Genetics of Spinal Muscular Atrophy: Progress and Challenges
Abstract
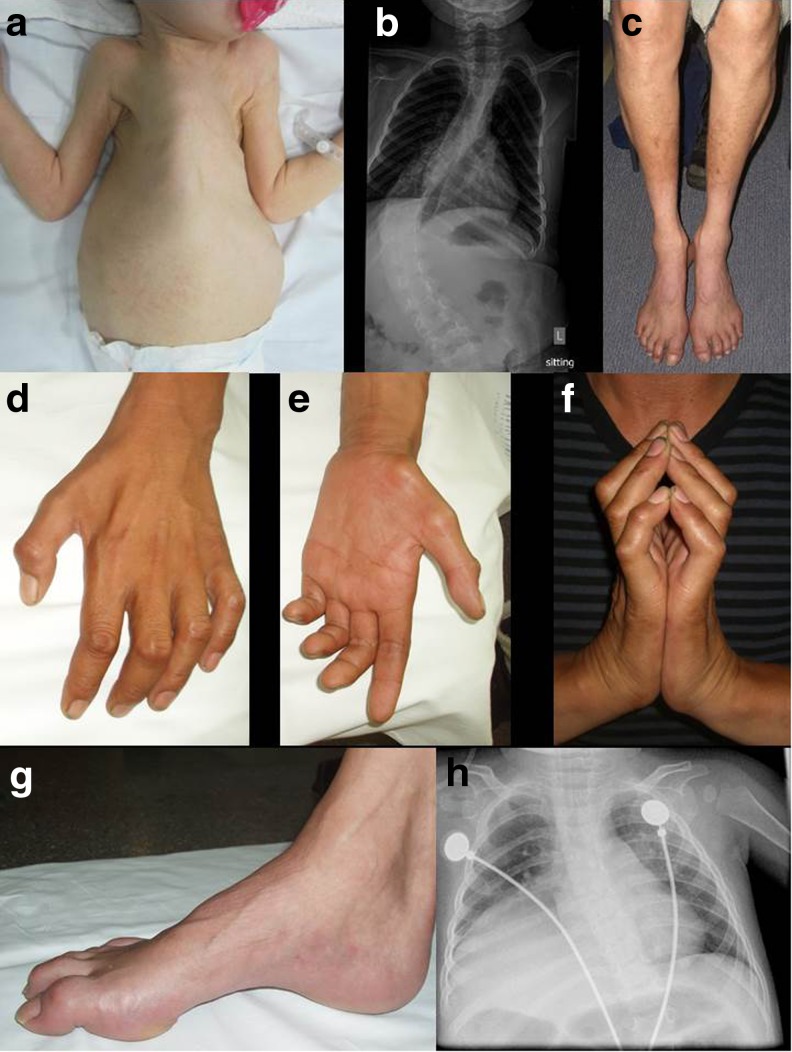
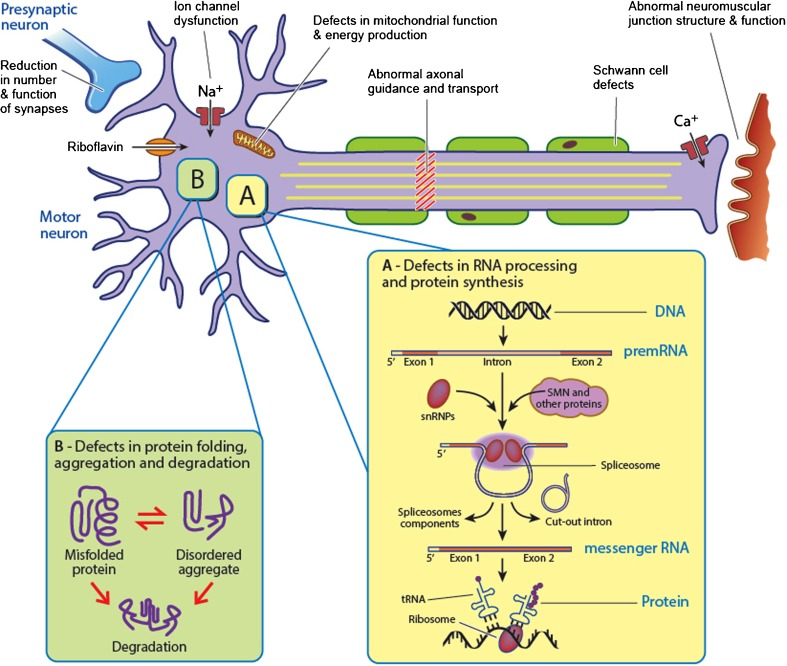
Spinal muscular atrophies (SMAs) are a group of inherited disorders characterized by motor neuron loss in the spinal cord and lower brainstem, muscle weakness, and atrophy. The clinical and genetic phenotypes incorporate a wide spectrum that is differentiated based on age of onset, pattern of muscle involvement, and inheritance pattern. Over the past several years, rapid advances in genetic technology have accelerated the identification of causative genes and provided important advances in understanding the molecular and biological basis of SMA and insights into the selective vulnerability of the motor neuron. Common pathophysiological themes include defects in RNA metabolism and splicing, axonal transport, and motor neuron development and connectivity. Together these have revealed potential novel treatment strategies, and extensive efforts are being undertaken towards expedited therapeutics. While a number of promising therapies for SMA are emerging, defining therapeutic windows and developing sensitive and relevant biomarkers are critical to facilitate potential success in clinical trials. This review incorporates an overview of the clinical manifestations and genetics of SMA, and describes recent advances in the understanding of mechanisms of disease pathogenesis and development of novel treatment strategies.
Figures
References
-
- Lefebvre S, Burglen L, Reboullet S, et al. Identification and characterization of a spinal muscular atrophy-determining gene. Cell. 1995;80:155–165. - PubMed
-
- Russman BS. Spinal muscular atrophy: clinical classification and disease heterogeneity. J Child Neurol. 2007;22:946–951. - PubMed
-
- Dressman D, Ahearn ME, Yariz KO, et al. X-linked infantile spinal muscular atrophy: clinical definition and molecular mapping. Genet Med. 2007;9:52–60. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
