

Large-scale mutational analysis of Kv11.1 reveals molecular insights into type 2 long QT syndrome
- PMID: 25417810
- PMCID: PMC4243539
- DOI: 10.1038/ncomms6535
Large-scale mutational analysis of Kv11.1 reveals molecular insights into type 2 long QT syndrome
Abstract
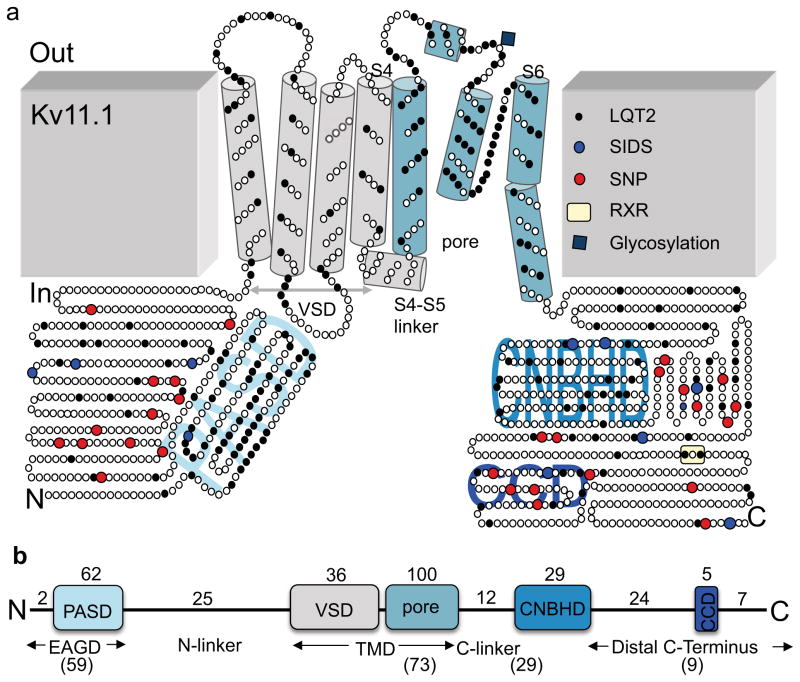
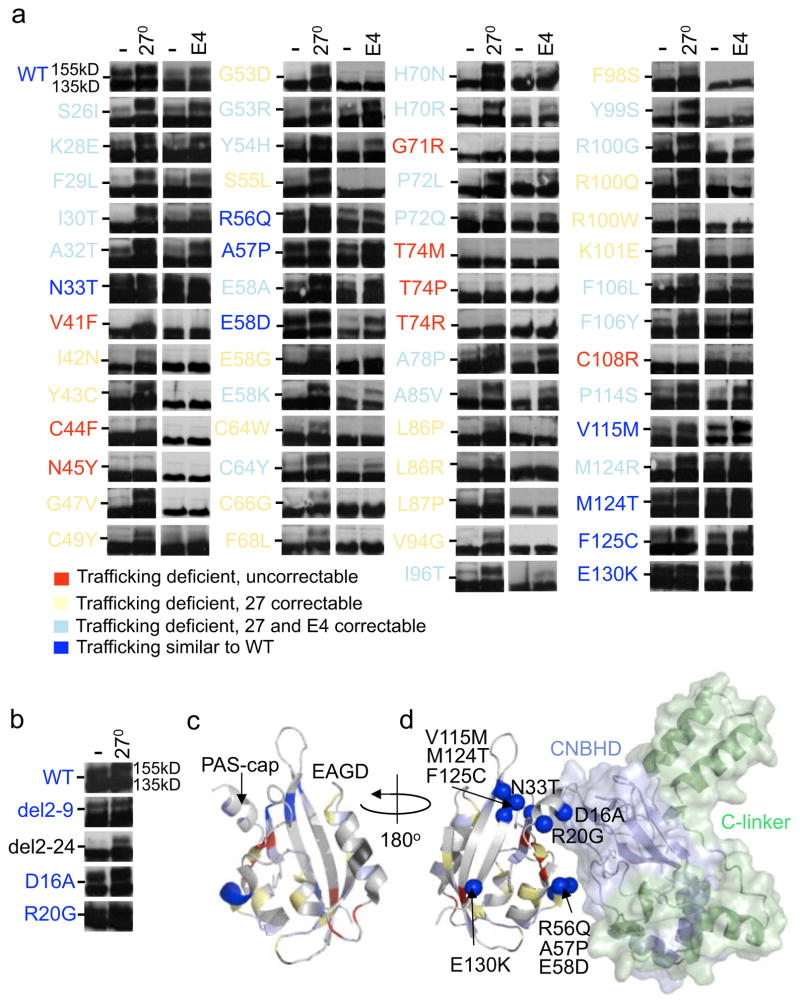
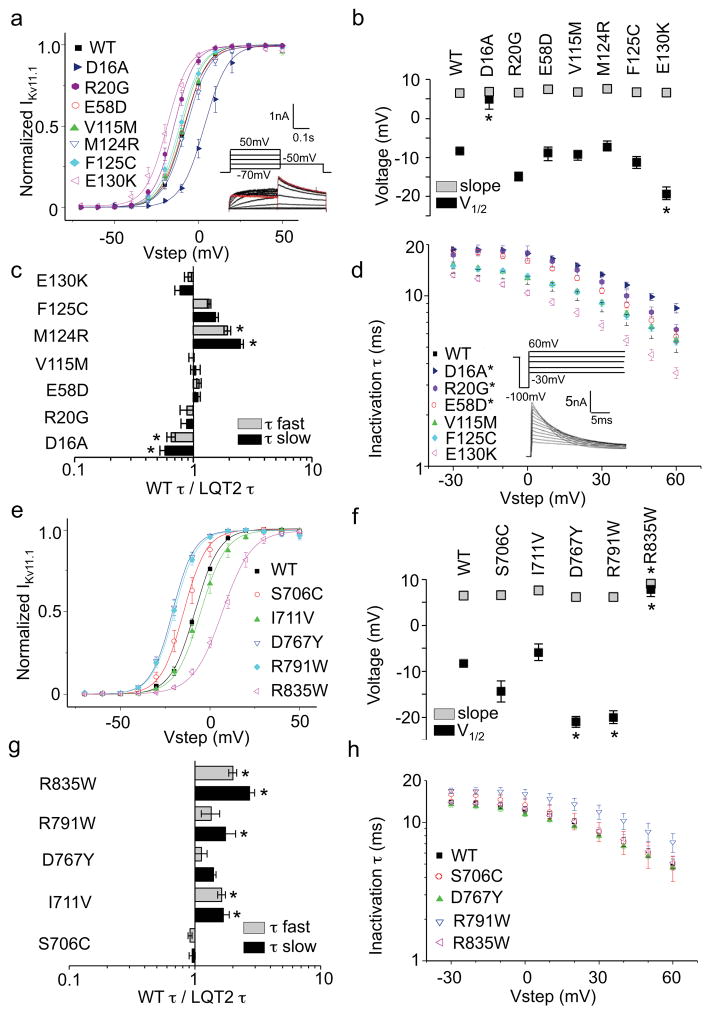
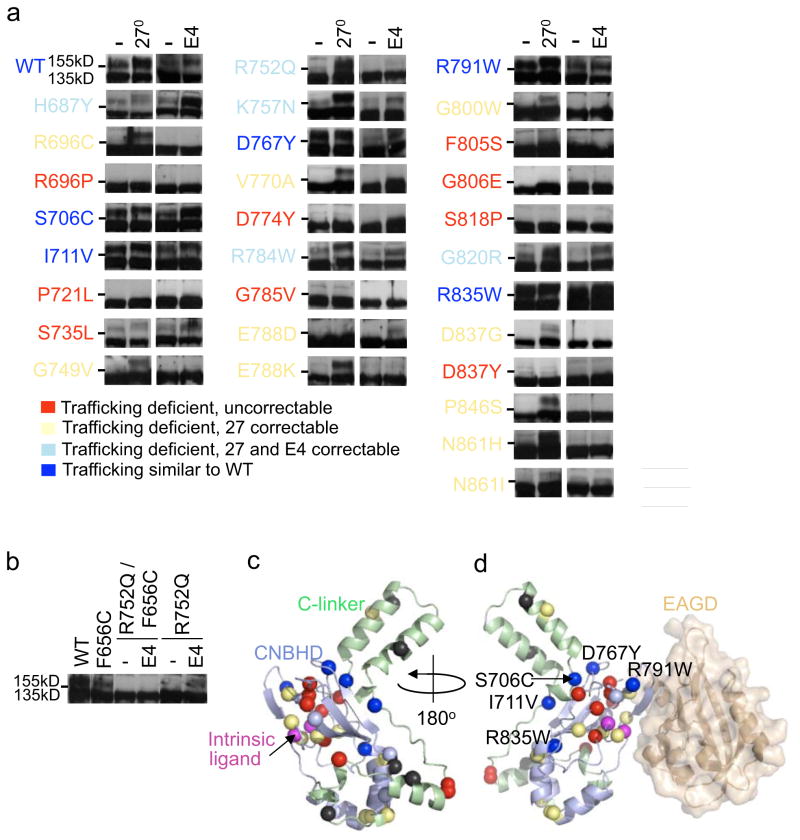
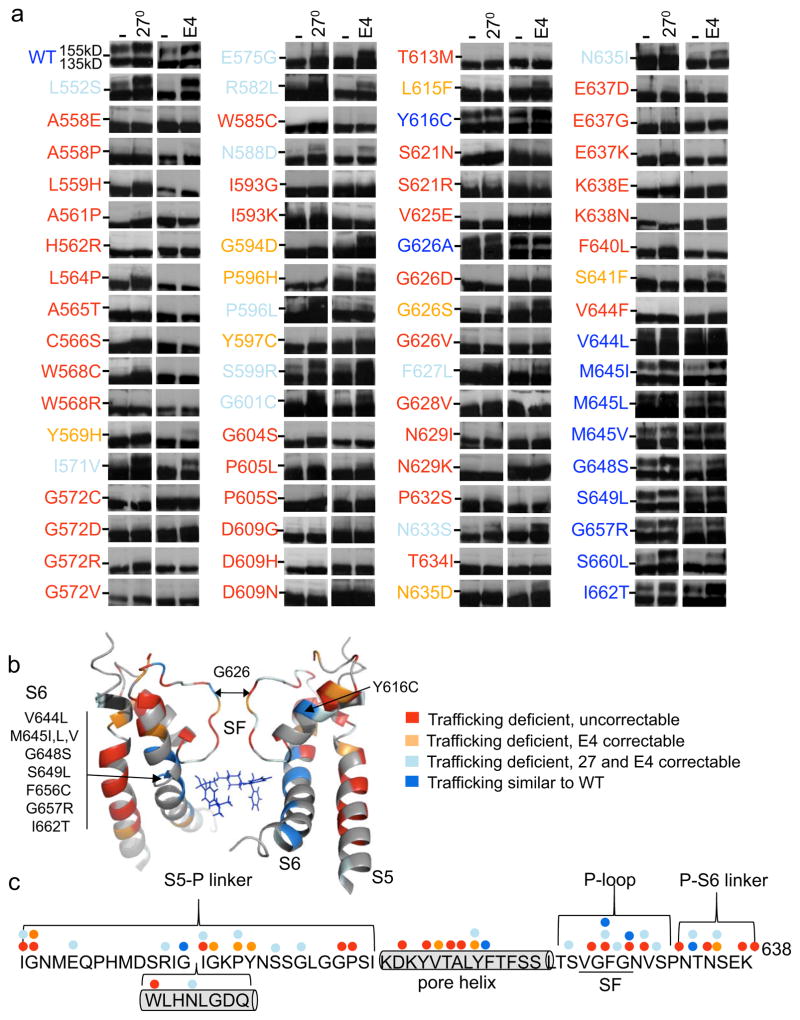
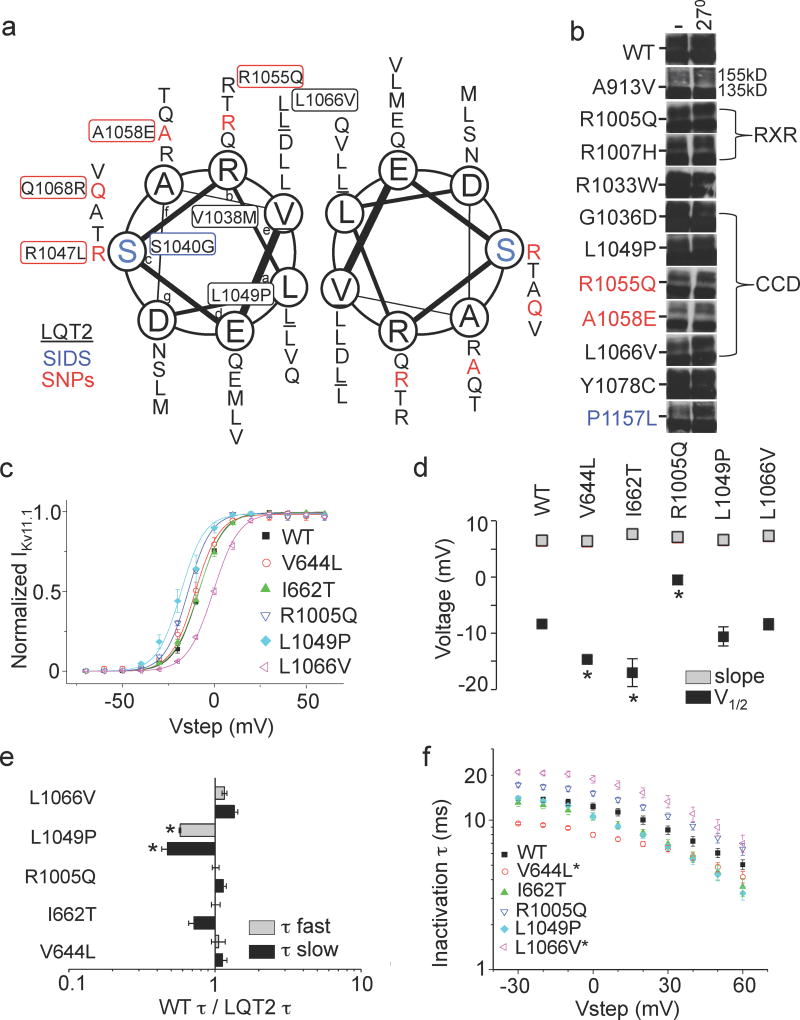
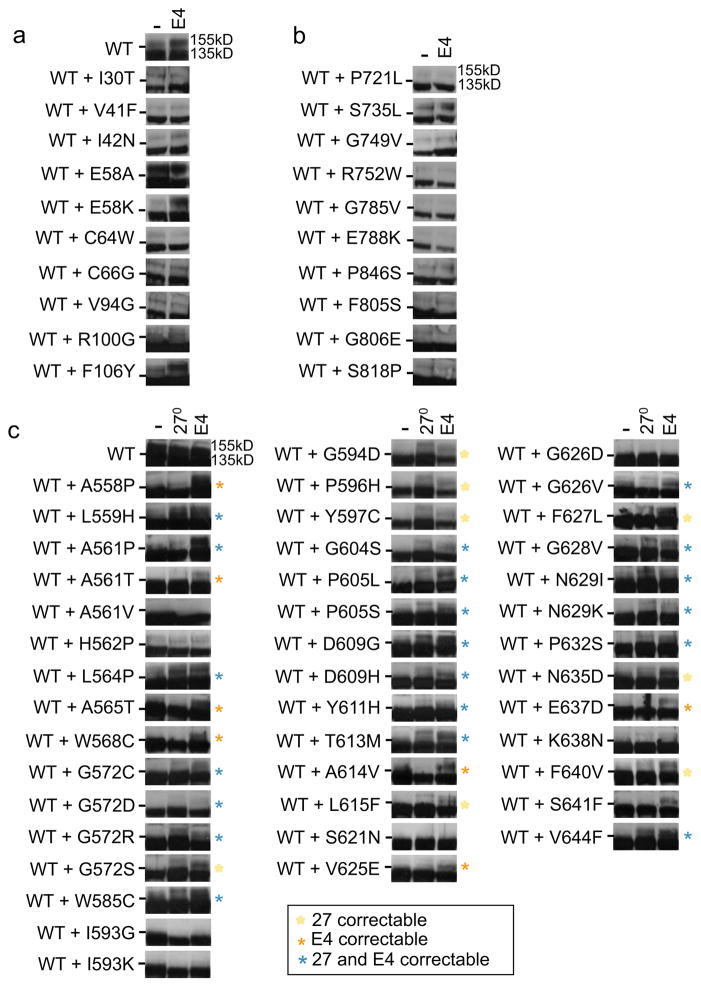
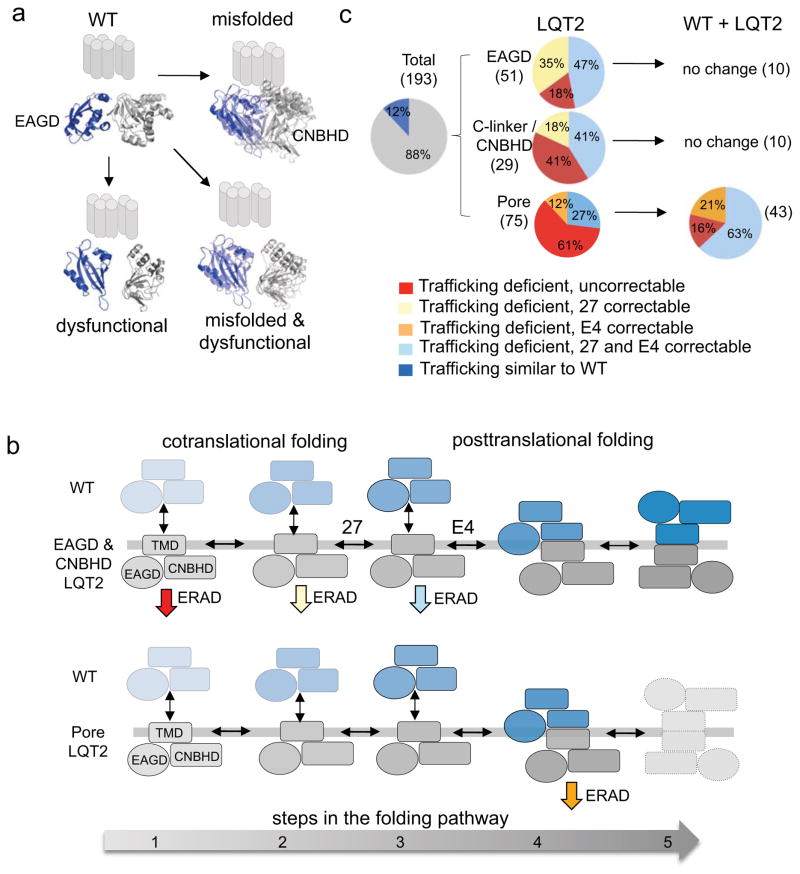
It has been suggested that deficient protein trafficking to the cell membrane is the dominant mechanism associated with type 2 Long QT syndrome (LQT2) caused by Kv11.1 potassium channel missense mutations, and that for many mutations the trafficking defect can be corrected pharmacologically. However, this inference was based on expression of a small number of Kv11.1 mutations. We performed a comprehensive analysis of 167 LQT2-linked missense mutations in four Kv11.1 structural domains and found that deficient protein trafficking is the dominant mechanism for all domains except for the distal carboxy-terminus. Also, most pore mutations--in contrast to intracellular domain mutations--were found to have severe dominant-negative effects when co-expressed with wild-type subunits. Finally, pharmacological correction of the trafficking defect in homomeric mutant channels was possible for mutations within all structural domains. However, pharmacological correction is dramatically improved for pore mutants when co-expressed with wild-type subunits to form heteromeric channels.
Conflict of interest statement
Figures

References
-
- Curran ME, et al. A molecular basis for cardiac arrhythmia: HERG mutations cause long QT syndrome. Cell. 1995;80(5):795–803. - PubMed
-
- Tester DJ, Ackerman MJ. Sudden infant death syndrome: How significant are the cardiac channelopathies? Cardiovasc Res. 2005;67:388–396. - PubMed
-
- Brugada R, et al. Sudden death associated with short-QT syndrome linked to mutations in HERG. Circulation. 2004;109(1):30–5. - PubMed
-
- Crociani O, et al. Cell cyce-dependent expression of HERG1 and HERG1B isoforms in tumor cells. J Biol Chem. 2003;278:2947–2955. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
