

Protein-protein docking: from interaction to interactome
- PMID: 25418159
- PMCID: PMC4213718
- DOI: 10.1016/j.bpj.2014.08.033
Protein-protein docking: from interaction to interactome
Abstract
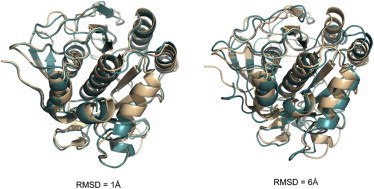
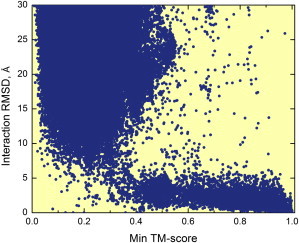
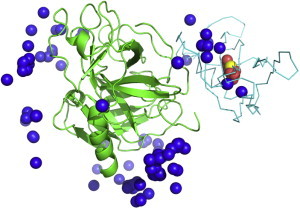
The protein-protein docking problem is one of the focal points of activity in computational biophysics and structural biology. The three-dimensional structure of a protein-protein complex, generally, is more difficult to determine experimentally than the structure of an individual protein. Adequate computational techniques to model protein interactions are important because of the growing number of known protein structures, particularly in the context of structural genomics. Docking offers tools for fundamental studies of protein interactions and provides a structural basis for drug design. Protein-protein docking is the prediction of the structure of the complex, given the structures of the individual proteins. In the heart of the docking methodology is the notion of steric and physicochemical complementarity at the protein-protein interface. Originally, mostly high-resolution, experimentally determined (primarily by x-ray crystallography) protein structures were considered for docking. However, more recently, the focus has been shifting toward lower-resolution modeled structures. Docking approaches have to deal with the conformational changes between unbound and bound structures, as well as the inaccuracies of the interacting modeled structures, often in a high-throughput mode needed for modeling of large networks of protein interactions. The growing number of docking developers is engaged in the community-wide assessments of predictive methodologies. The development of more powerful and adequate docking approaches is facilitated by rapidly expanding information and data resources, growing computational capabilities, and a deeper understanding of the fundamental principles of protein interactions.
Figures


References
-
- Platzer K.E.B., Momany F.A., Scheraga H.A. Conformational energy calculations of enzyme-substrate interactions. II. Computation of the binding energy for substrates in the active site of alpha-chymotrypsin. Int. J. Pept. Protein Res. 1972;4:201–219. - PubMed
-
- Wodak S.Y., Liu M.Y., Wyckoff H.W. The structure of cytidilyl(2′,5′)adenosine when bound to pancreatic ribonuclease S. J. Mol. Biol. 1977;116:855–875. - PubMed
-
- Pincus M.R., Scheraga H.A. Conformational energy calculations of enzyme-substrate and enzyme-inhibitor complexes of lysozyme. 2. Calculation of the structures of complexes with flexible enzyme. Macromolecules. 1979;12:633–644.
-
- Wodak S.J., Janin J. Computer analysis of protein-protein interaction. J. Mol. Biol. 1978;124:323–342. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
