

Transmission characteristics of variably protease-sensitive prionopathy
- PMID: 25418590
- PMCID: PMC4257788
- DOI: 10.3201/eid2012.140548
Transmission characteristics of variably protease-sensitive prionopathy
Abstract
Variably protease-sensitive prionopathy (VPSPr), a recently identified and seemingly sporadic human prion disease, is distinct from Creutzfeldt-Jakob disease (CJD) but shares features of Gerstmann-Sträussler-Scheinker disease (GSS). However, contrary to exclusively inherited GSS, no prion protein (PrP) gene variations have been detected in VPSPr, suggesting that VPSPr might be the long-sought sporadic form of GSS. The VPSPr atypical features raised the issue of transmissibility, a prototypical property of prion diseases. We inoculated VPSPr brain homogenate into transgenic mice expressing various levels of human PrP (PrPC). On first passage, 54% of challenged mice showed histopathologic lesions, and 34% harbored abnormal PrP similar to that of VPSPr. Surprisingly, no prion disease was detected on second passage. We concluded that VPSPr is transmissible; thus, it is an authentic prion disease. However, we speculate that normal human PrPC is not an efficient conversion substrate (or mouse brain not a favorable environment) and therefore cannot sustain replication beyond the first passage.
Figures
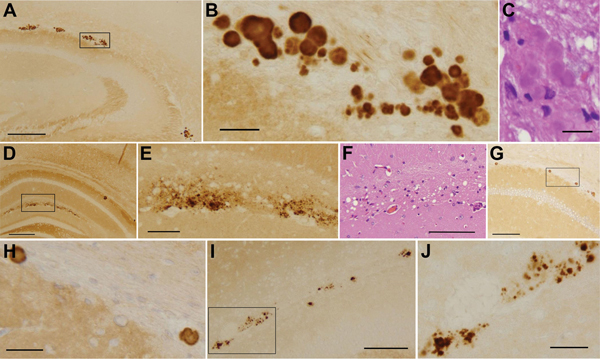
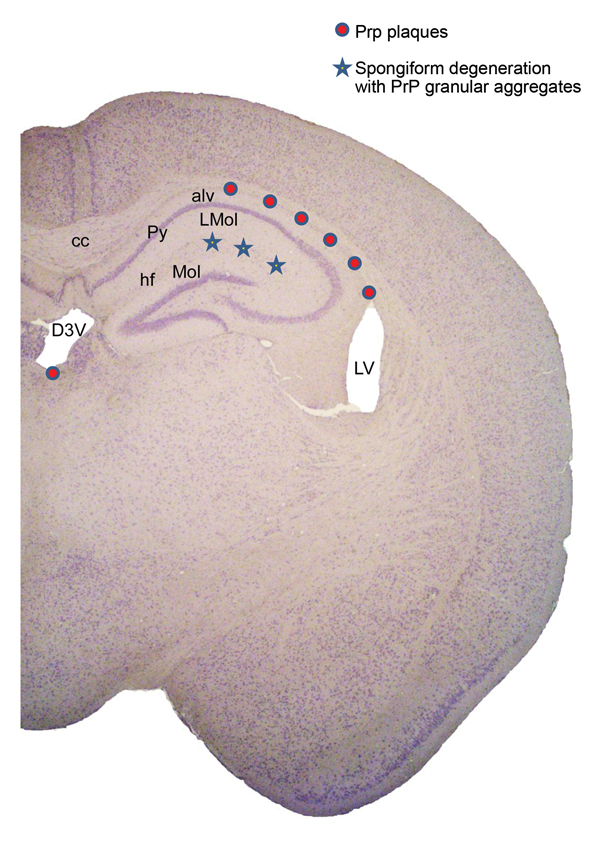
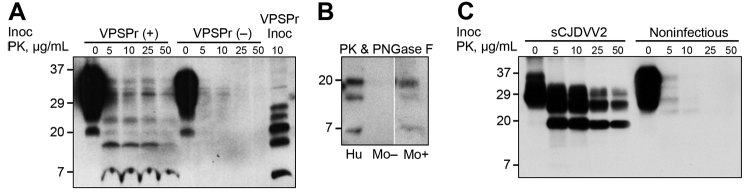
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
