

Chronic enrichment of hepatic endoplasmic reticulum-mitochondria contact leads to mitochondrial dysfunction in obesity
- PMID: 25419710
- PMCID: PMC4412031
- DOI: 10.1038/nm.3735
Chronic enrichment of hepatic endoplasmic reticulum-mitochondria contact leads to mitochondrial dysfunction in obesity
Abstract
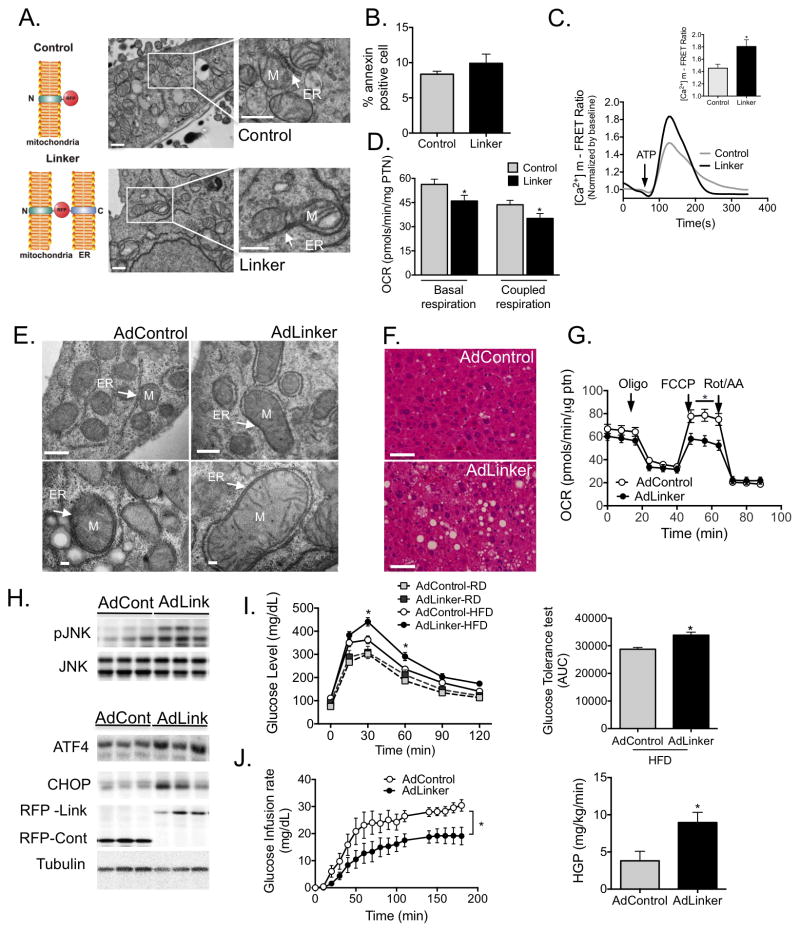
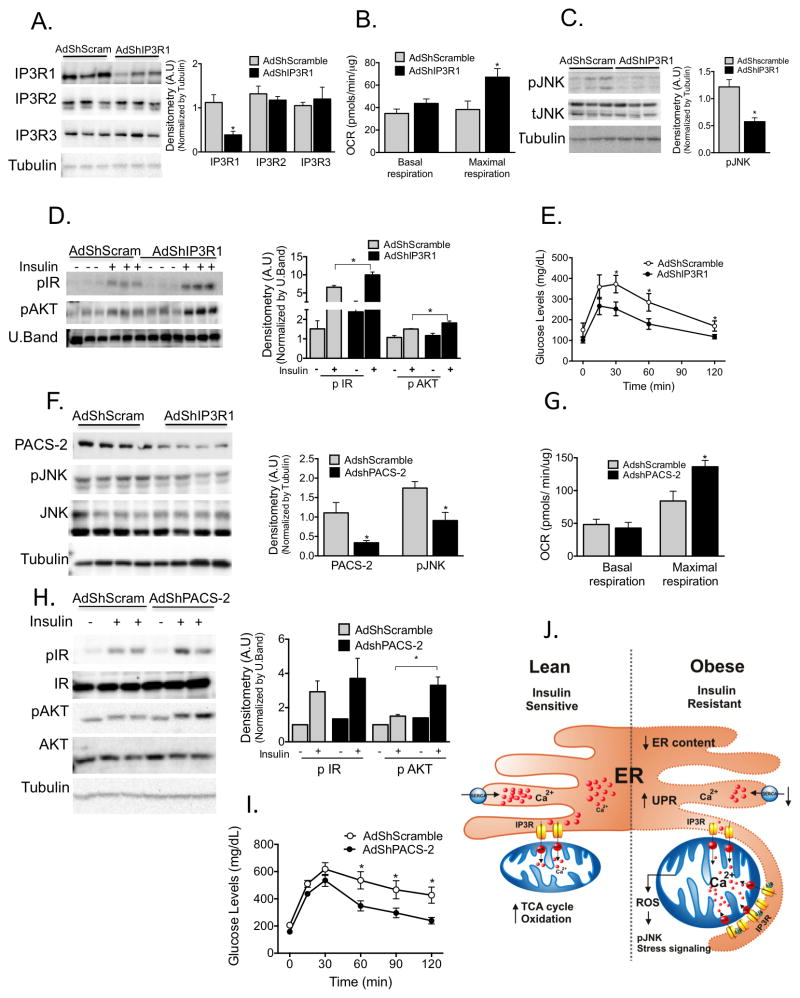
Proper function of the endoplasmic reticulum (ER) and mitochondria is crucial for cellular homeostasis, and dysfunction at either site has been linked to pathophysiological states, including metabolic diseases. Although the ER and mitochondria play distinct cellular roles, these organelles also form physical interactions with each other at sites defined as mitochondria-associated ER membranes (MAMs), which are essential for calcium, lipid and metabolite exchange. Here we show that in the liver, obesity leads to a marked reorganization of MAMs resulting in mitochondrial calcium overload, compromised mitochondrial oxidative capacity and augmented oxidative stress. Experimental induction of ER-mitochondria interactions results in oxidative stress and impaired metabolic homeostasis, whereas downregulation of PACS-2 or IP3R1, proteins important for ER-mitochondria tethering or calcium transport, respectively, improves mitochondrial oxidative capacity and glucose metabolism in obese animals. These findings establish excessive ER-mitochondrial coupling as an essential component of organelle dysfunction in obesity that may contribute to the development of metabolic pathologies such as insulin resistance and diabetes.
Figures
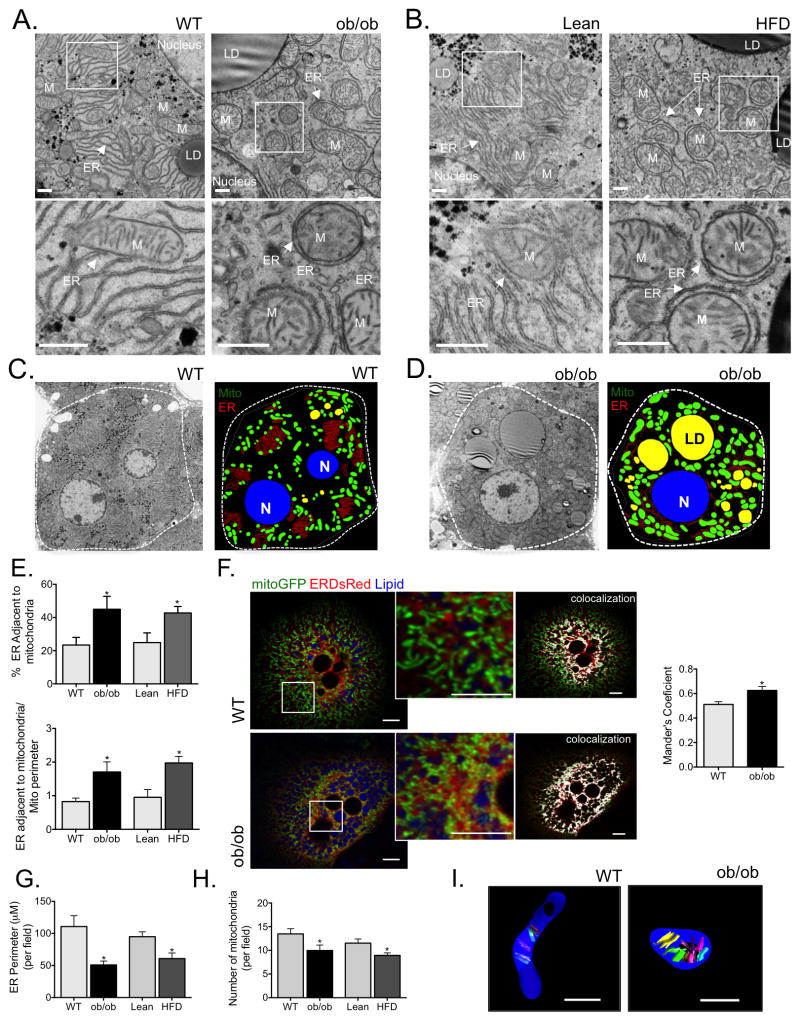
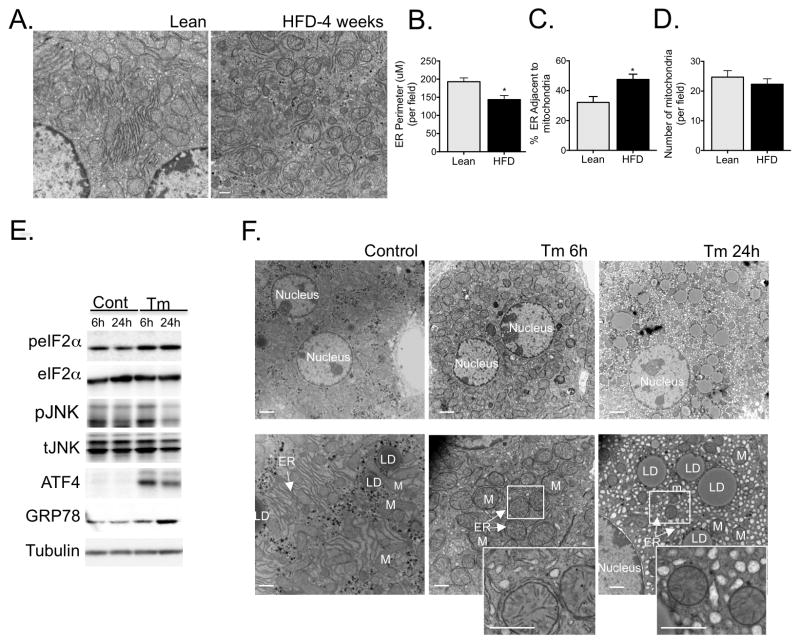
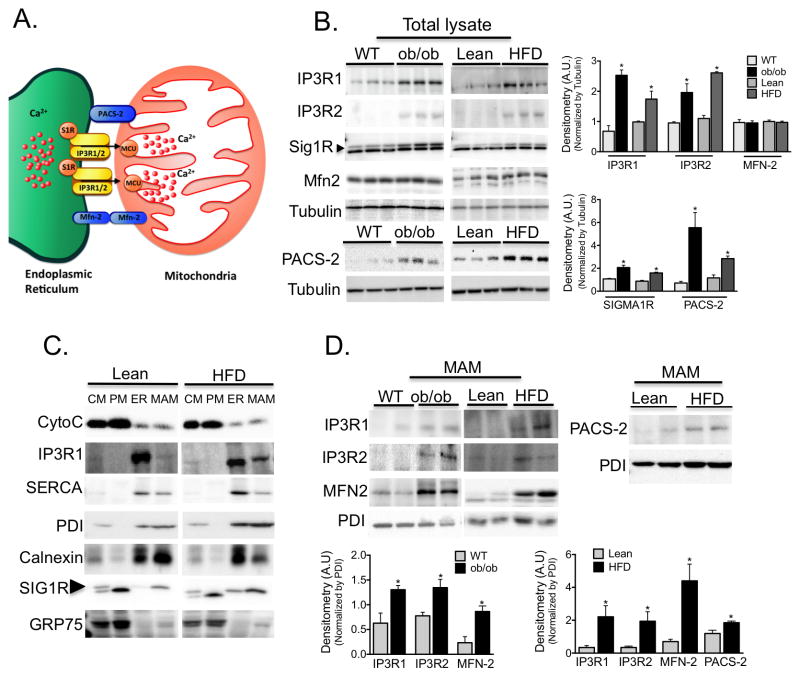
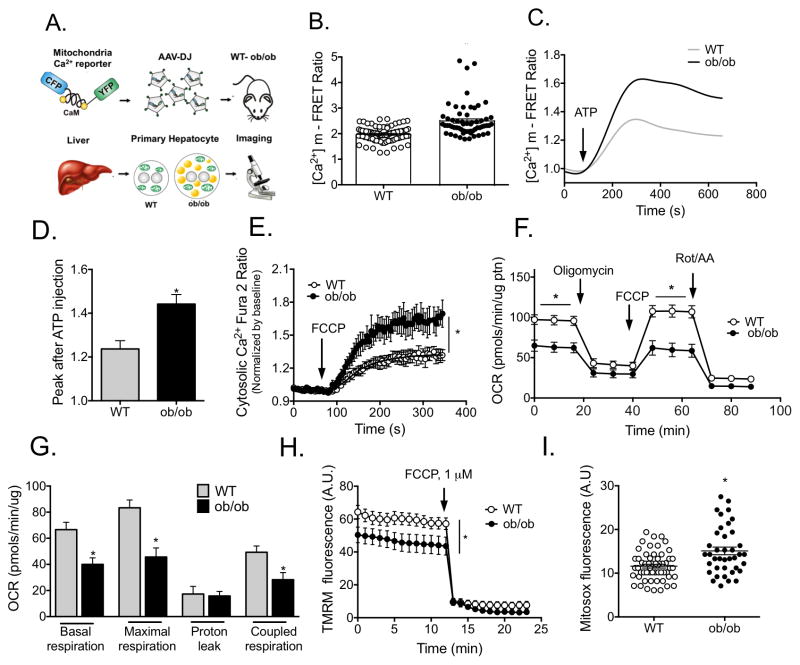
References
-
- Flegal KM, Carroll MD, Ogden CL, Curtin LR. Prevalence and trends in obesity among US adults, 1999–2008. JAMA: the journal of the American Medical Association. 2010;303:235–241. - PubMed
-
- Hotamisligil GS. Inflammation and metabolic disorders. Nature. 2006;444:860–867. - PubMed
-
- Lowell BB, Shulman GI. Mitochondrial dysfunction and type 2 diabetes. Science. 2005;307:384–387. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
