

Regulatory network decoded from epigenomes of surface ectoderm-derived cell types
- PMID: 25421844
- PMCID: PMC4385735
- DOI: 10.1038/ncomms6442
Regulatory network decoded from epigenomes of surface ectoderm-derived cell types
Abstract
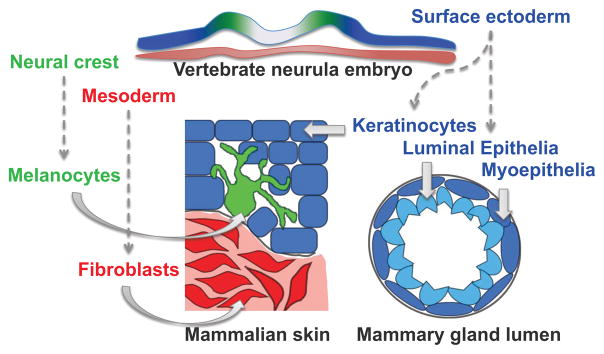
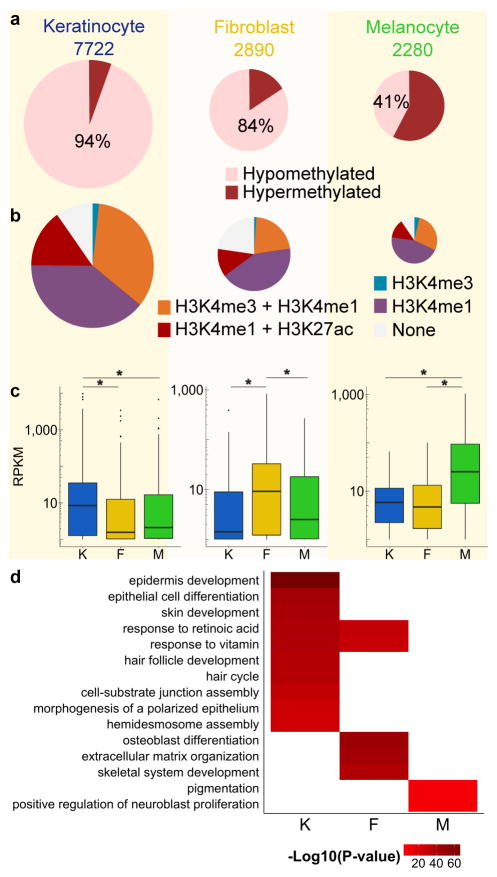
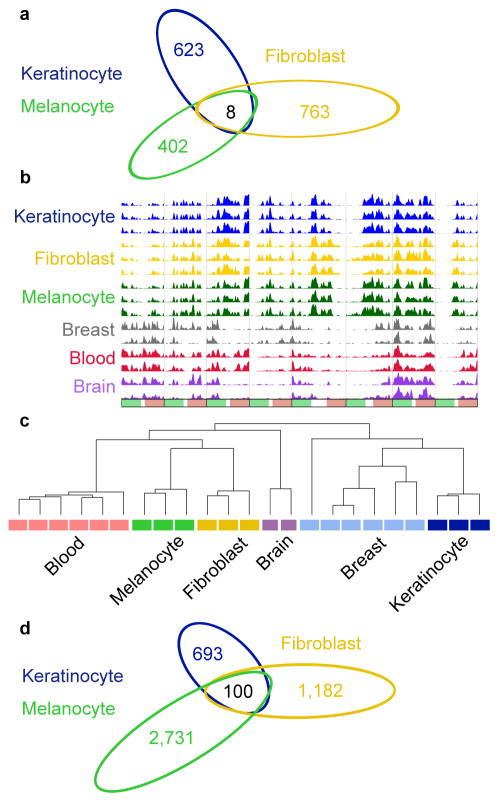
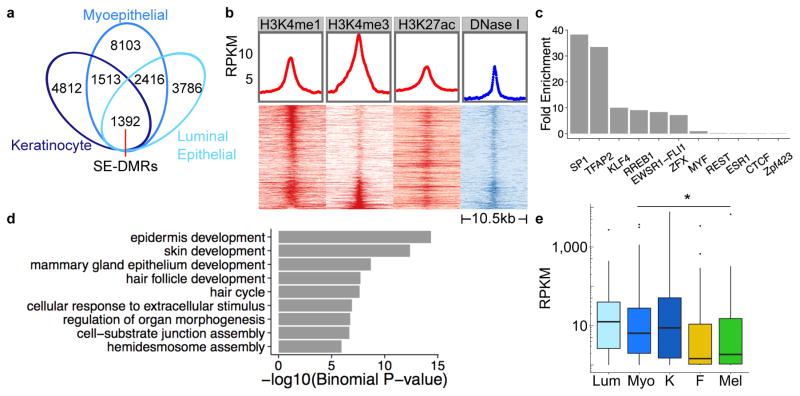
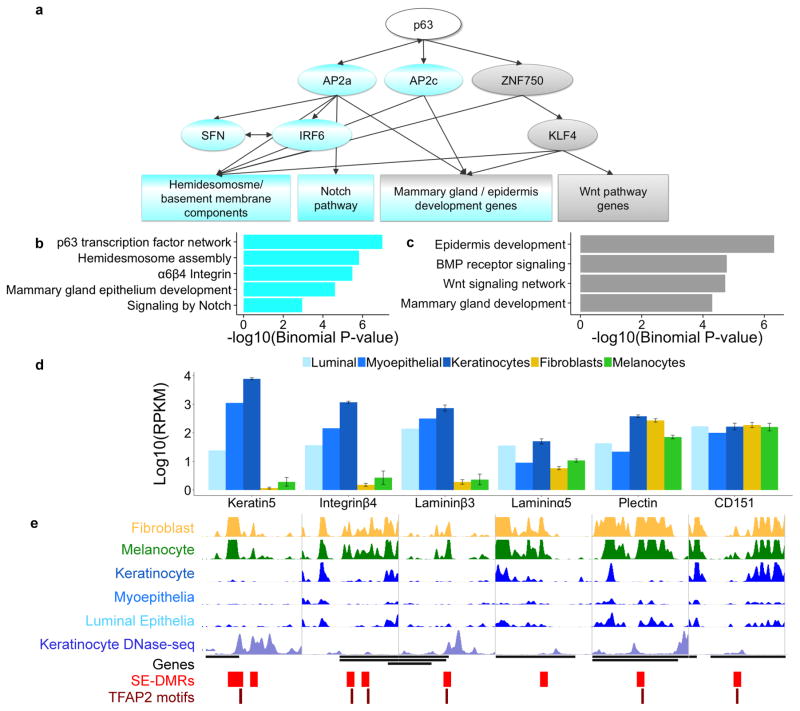
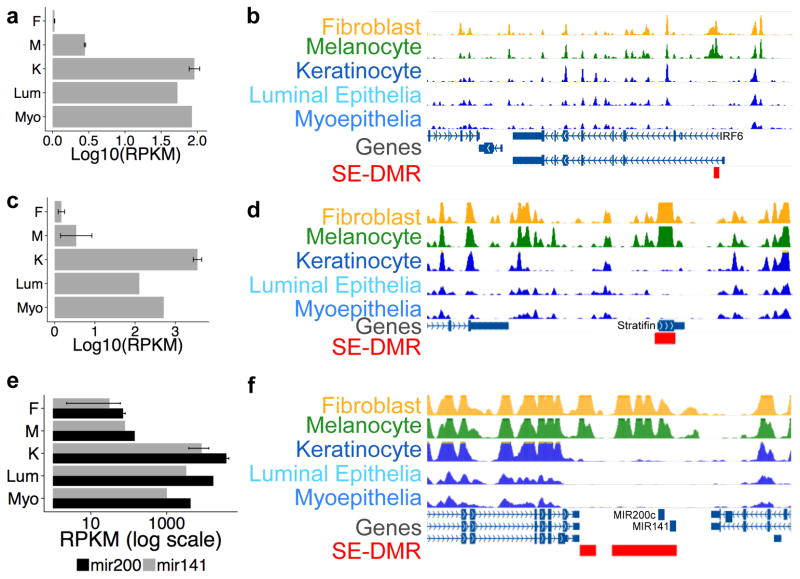
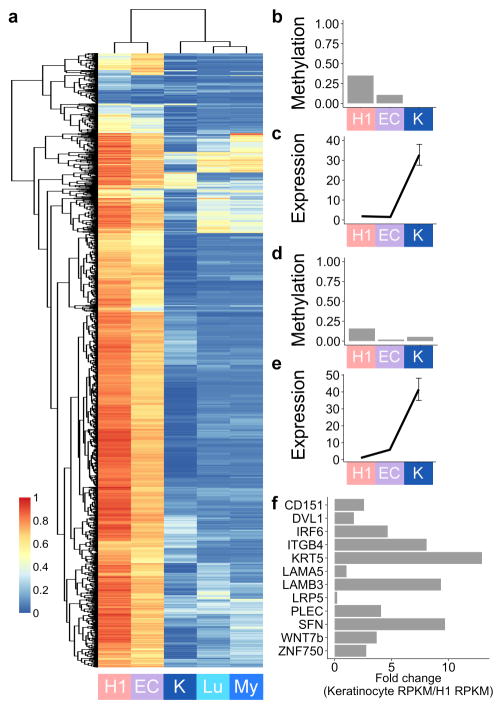
Developmental history shapes the epigenome and biological function of differentiated cells. Epigenomic patterns have been broadly attributed to the three embryonic germ layers. Here we investigate how developmental origin influences epigenomes. We compare key epigenomes of cell types derived from surface ectoderm (SE), including keratinocytes and breast luminal and myoepithelial cells, against neural crest-derived melanocytes and mesoderm-derived dermal fibroblasts, to identify SE differentially methylated regions (SE-DMRs). DNA methylomes of neonatal keratinocytes share many more DMRs with adult breast luminal and myoepithelial cells than with melanocytes and fibroblasts from the same neonatal skin. This suggests that SE origin contributes to DNA methylation patterning, while shared skin tissue environment has limited effect on epidermal keratinocytes. Hypomethylated SE-DMRs are in proximity to genes with SE relevant functions. They are also enriched for enhancer- and promoter-associated histone modifications in SE-derived cells, and for binding motifs of transcription factors important in keratinocyte and mammary gland biology. Thus, epigenomic analysis of cell types with common developmental origin reveals an epigenetic signature that underlies a shared gene regulatory network.
Conflict of interest statement
The authors declare no competing financial interests.
Figures
References
Publication types
MeSH terms
Associated data
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
Grants and funding
- 5U01ES017154/ES/NIEHS NIH HHS/United States
- U01 ES017154/ES/NIEHS NIH HHS/United States
- R01HG007354/HG/NHGRI NIH HHS/United States
- R01 HG007175/HG/NHGRI NIH HHS/United States
- R01HG007175/HG/NHGRI NIH HHS/United States
- 5T32GM081739/GM/NIGMS NIH HHS/United States
- R01 ES024992/ES/NIEHS NIH HHS/United States
- T32 GM081739/GM/NIGMS NIH HHS/United States
- R25 DA027995/DA/NIDA NIH HHS/United States
- R01AG028492/AG/NIA NIH HHS/United States
- R01 HG007354/HG/NHGRI NIH HHS/United States
- R01 AG028492/AG/NIA NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
