

Crystal structure of the sodium-proton antiporter NhaA dimer and new mechanistic insights
- PMID: 25422503
- PMCID: PMC4242812
- DOI: 10.1085/jgp.201411219
Crystal structure of the sodium-proton antiporter NhaA dimer and new mechanistic insights
Abstract
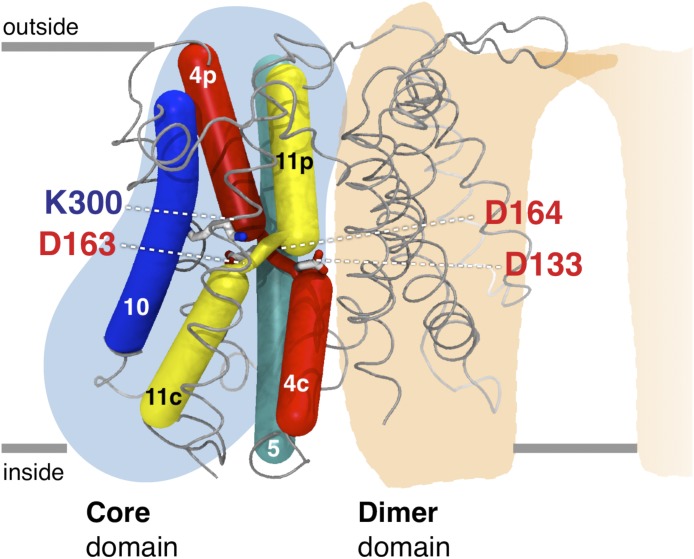
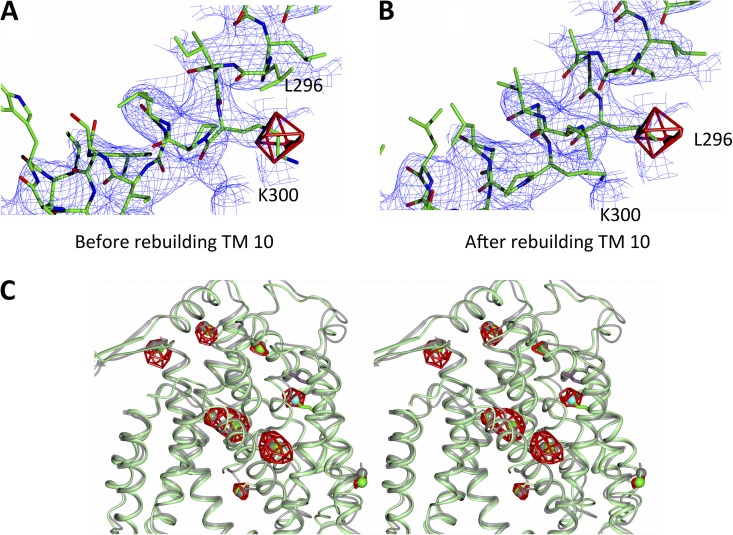
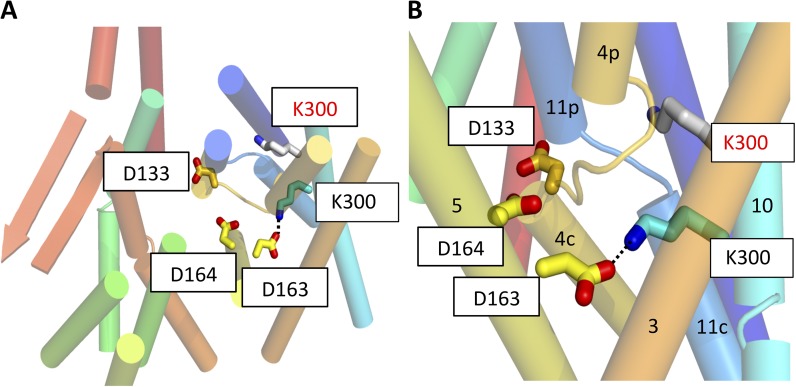
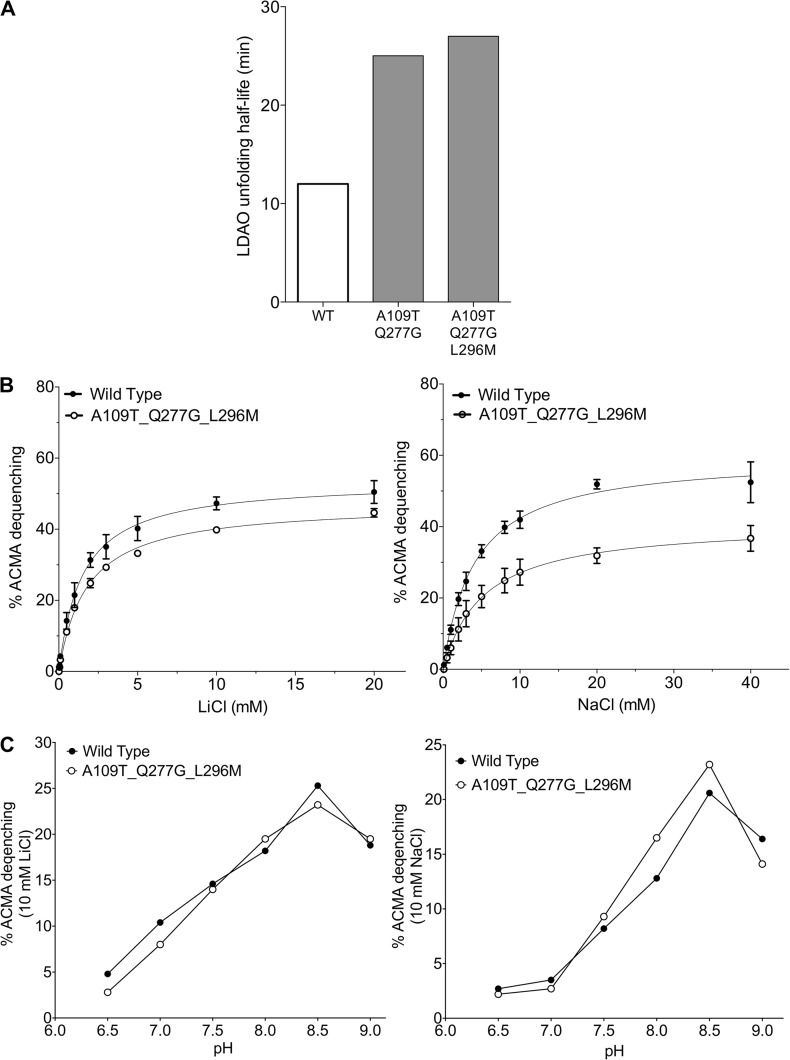
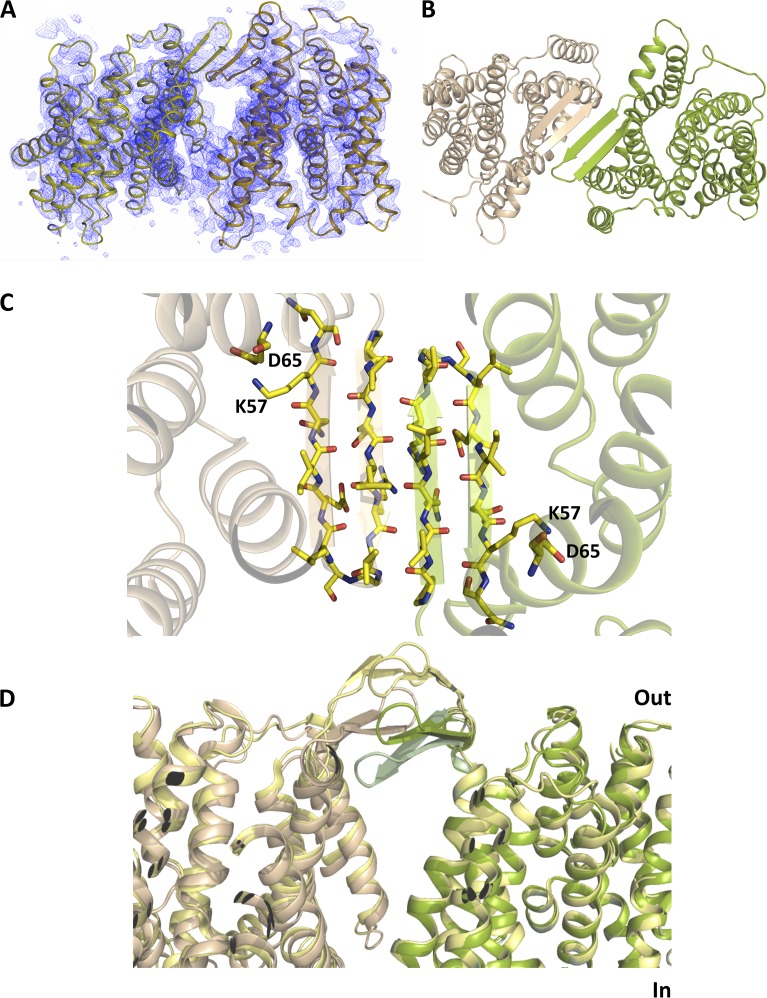
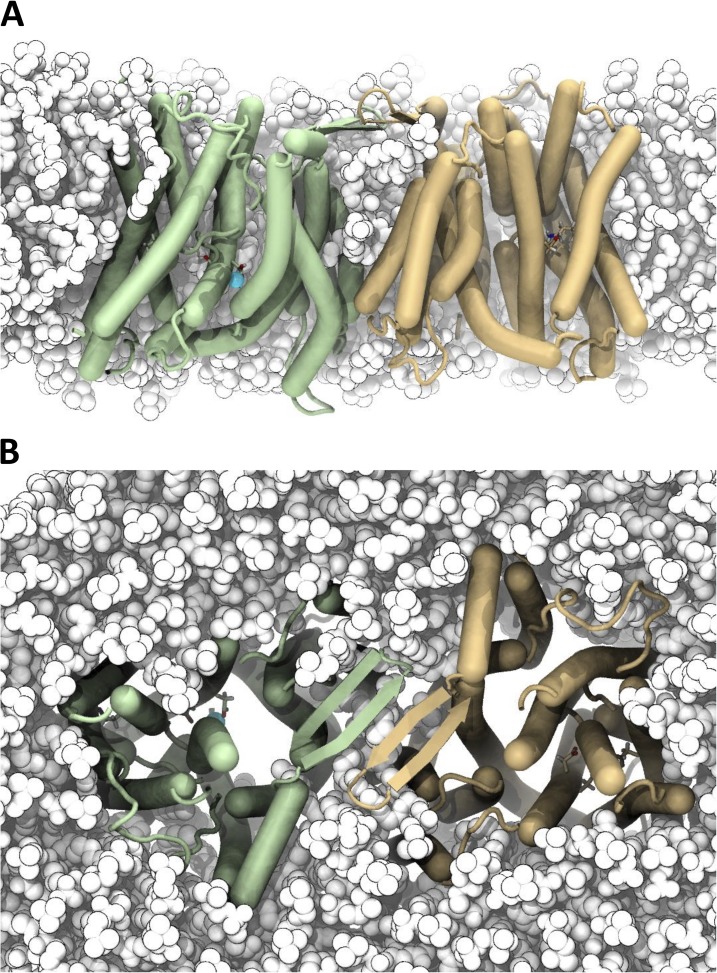
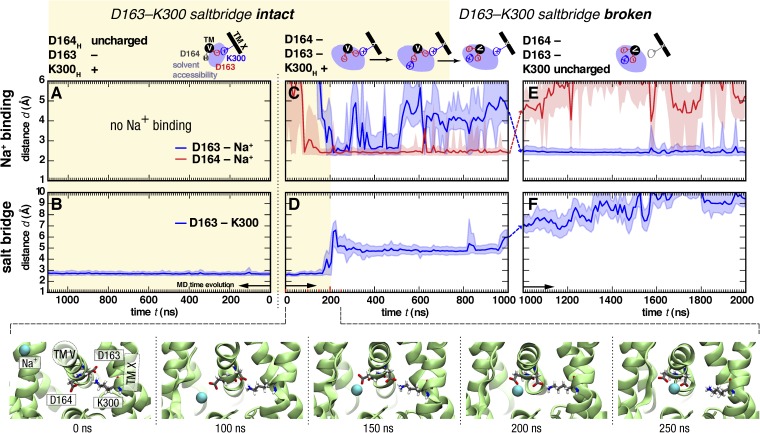
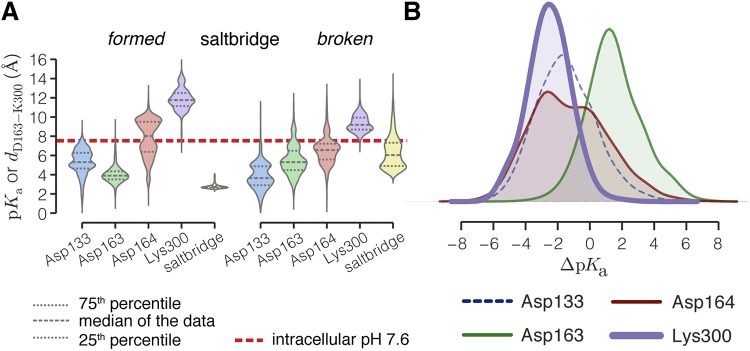
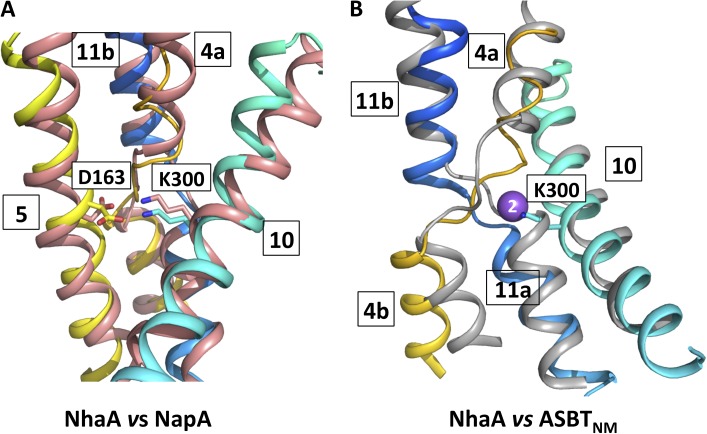
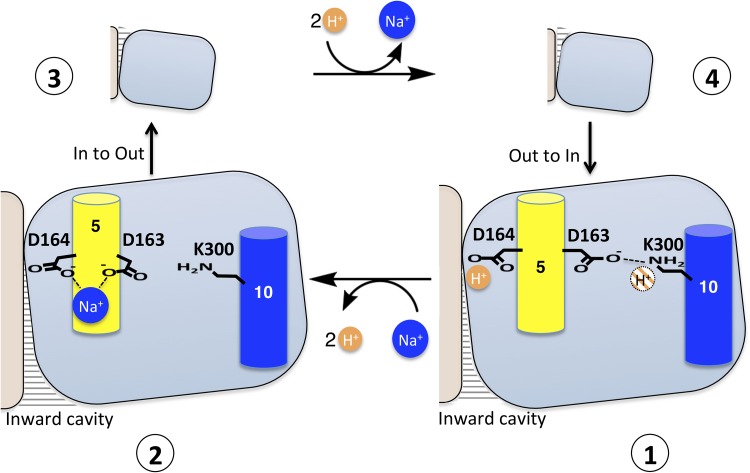
Sodium-proton antiporters rapidly exchange protons and sodium ions across the membrane to regulate intracellular pH, cell volume, and sodium concentration. How ion binding and release is coupled to the conformational changes associated with transport is not clear. Here, we report a crystal form of the prototypical sodium-proton antiporter NhaA from Escherichia coli in which the protein is seen as a dimer. In this new structure, we observe a salt bridge between an essential aspartic acid (Asp163) and a conserved lysine (Lys300). An equivalent salt bridge is present in the homologous transporter NapA, but not in the only other known crystal structure of NhaA, which provides the foundation of most existing structural models of electrogenic sodium-proton antiport. Molecular dynamics simulations show that the stability of the salt bridge is weakened by sodium ions binding to Asp164 and the neighboring Asp163. This suggests that the transport mechanism involves Asp163 switching between forming a salt bridge with Lys300 and interacting with the sodium ion. pKa calculations suggest that Asp163 is highly unlikely to be protonated when involved in the salt bridge. As it has been previously suggested that Asp163 is one of the two residues through which proton transport occurs, these results have clear implications to the current mechanistic models of sodium-proton antiport in NhaA.
© 2014 Lee et al.
Figures
References
-
- Adams P.D., Afonine P.V., Bunkóczi G., Chen V.B., Davis I.W., Echols N., Headd J.J., Hung L.W., Kapral G.J., Grosse-Kunstleve R.W., et al. . 2010. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66:213–221 10.1107/S0907444909052925 - DOI - PMC - PubMed
-
- Berendsen H.J.C., Postma J.P.M., van Gunsteren W.F., DiNola A., and Haak J.R.. 1984. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 81:3684–3690 10.1063/1.448118 - DOI
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
