

SRST2: Rapid genomic surveillance for public health and hospital microbiology labs
- PMID: 25422674
- PMCID: PMC4237778
- DOI: 10.1186/s13073-014-0090-6
SRST2: Rapid genomic surveillance for public health and hospital microbiology labs
Abstract
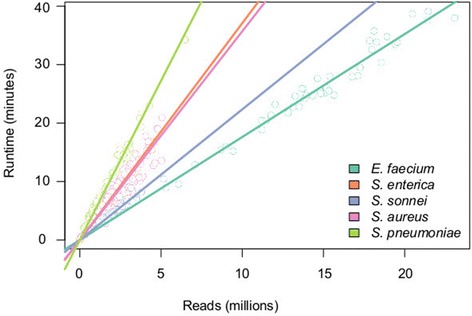
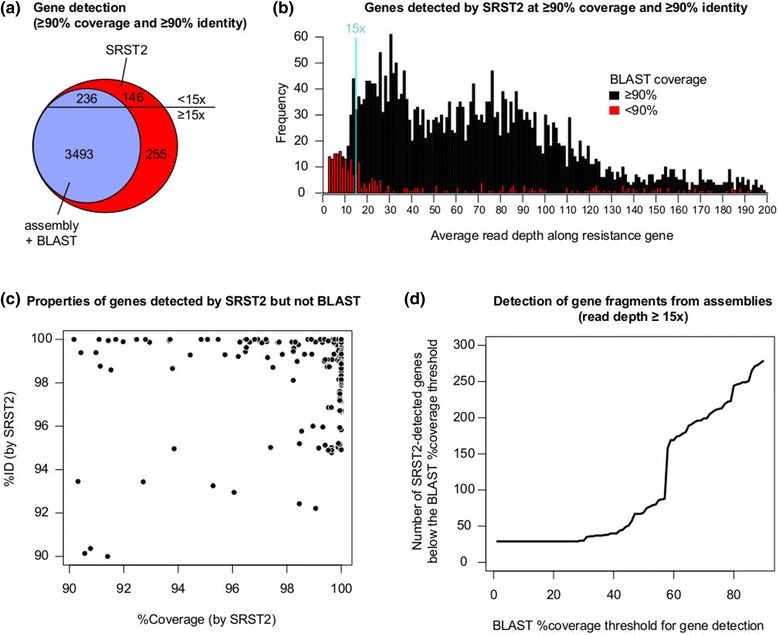
Rapid molecular typing of bacterial pathogens is critical for public health epidemiology, surveillance and infection control, yet routine use of whole genome sequencing (WGS) for these purposes poses significant challenges. Here we present SRST2, a read mapping-based tool for fast and accurate detection of genes, alleles and multi-locus sequence types (MLST) from WGS data. Using >900 genomes from common pathogens, we show SRST2 is highly accurate and outperforms assembly-based methods in terms of both gene detection and allele assignment. We include validation of SRST2 within a public health laboratory, and demonstrate its use for microbial genome surveillance in the hospital setting. In the face of rising threats of antimicrobial resistance and emerging virulence among bacterial pathogens, SRST2 represents a powerful tool for rapidly extracting clinically useful information from raw WGS data. Source code is available from http://katholt.github.io/srst2/.
Figures






References
-
- Sabat AJ, Budimir A, Nashev D, Sa-Leao R, van Dijl J, Laurent F, Grundmann H, Friedrich AW, Markers ESGoE: Overview of molecular typing methods for outbreak detection and epidemiological surveillance.Euro Surveill 2013, 18:20380. - PubMed
LinkOut - more resources
Full Text Sources
Other Literature Sources
