

Cryptogenic stroke and small fiber neuropathy of unknown etiology in patients with alpha-galactosidase A -10T genotype
- PMID: 25423912
- PMCID: PMC4255940
- DOI: 10.1186/s13023-014-0178-5
Cryptogenic stroke and small fiber neuropathy of unknown etiology in patients with alpha-galactosidase A -10T genotype
Abstract
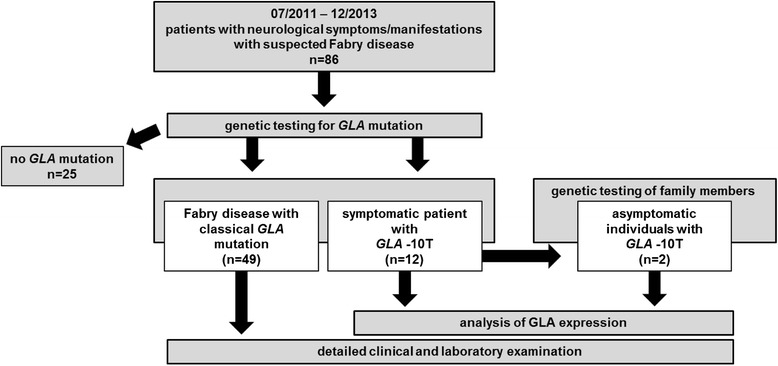
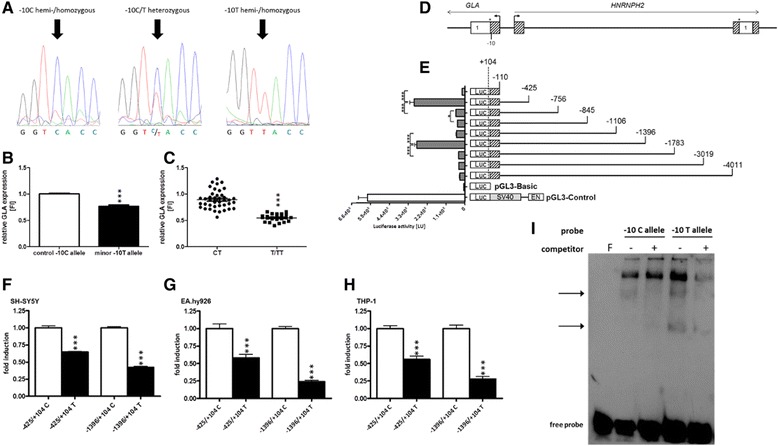
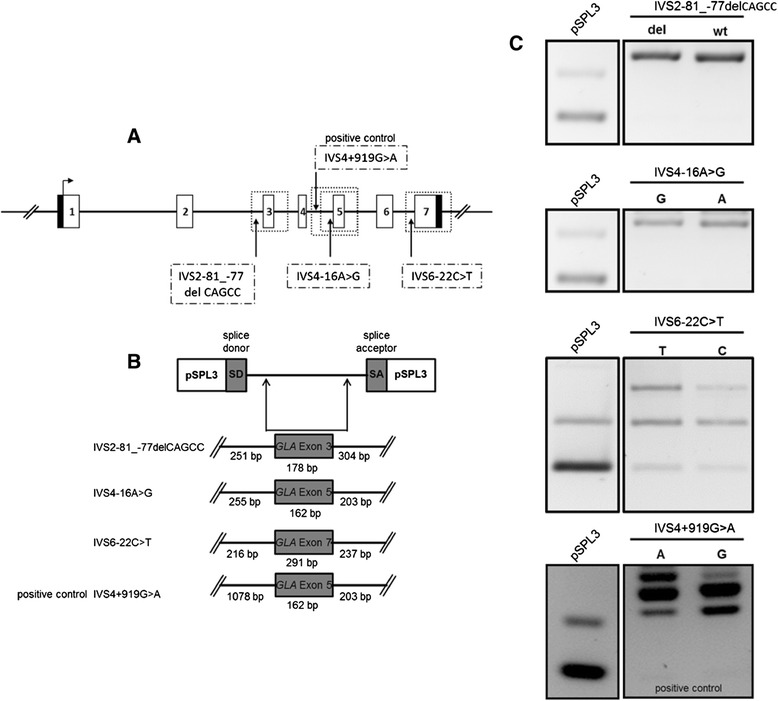
Background: Fabry disease (FD) is a multisystemic disorder with typical neurological manifestations such as stroke and small fiber neuropathy (SFN), caused by mutations of the alpha-galactosidase A (GLA) gene. We analyzed 15 patients carrying the GLA haplotype -10C>T [rs2071225], IVS2-81_-77delCAGCC [rs5903184], IVS4-16A>G [rs2071397], and IVS6-22C>T [rs2071228] for potential neurological manifestations.
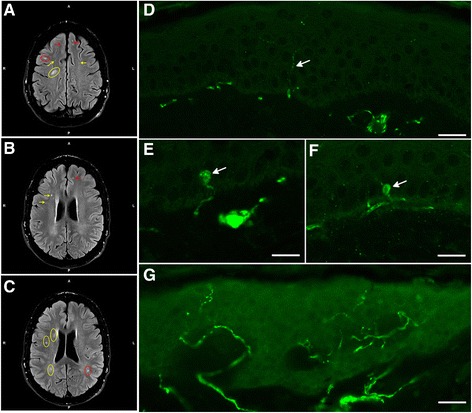
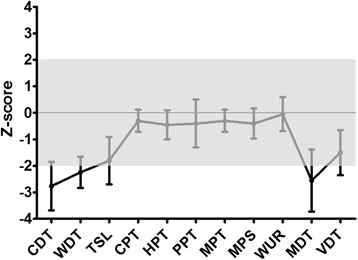
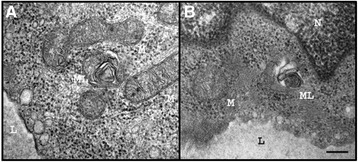
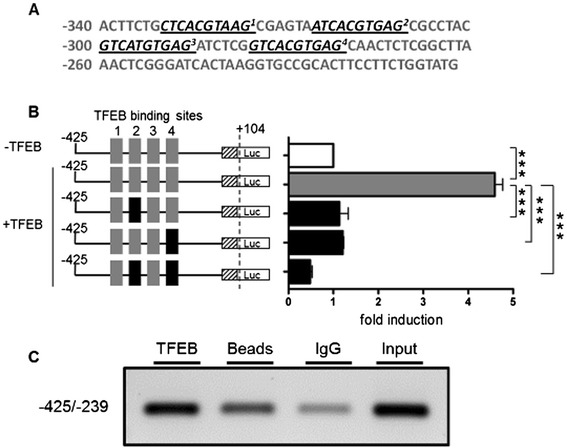
Methods and results: Patients were retrospectively analyzed for stroke, transient ischemic attack (TIA), white matter lesions (WML) and SFN with neuropathic pain. Functional impact of the haplotype was determined by molecular genetic methods including real-time PCR, exon trapping, promoter deletion constructs and electrophoretic mobility shift assays. Symptomatic -10T allele carriers suffered from stroke, TIA, WML, and SFN with neuropathic pain. Patients' mean GLA mRNA expression level was reduced to ~70% (p < 0.0001) and a dose-dependent effect of the -10T allele on GLA mRNA expression was observed in hemi/homozygous compared to heterozygous patients (p < 0.0001). Molecular analyzes revealed that the -10T allele resulted in a reduced promoter activity and an altered transcription factor binding, while a functional relevance of the co-segregated intronic variants was excluded by exon trapping.
Conclusions: Based on this complementary approach of clinical observation and functional testing, we conclude that the GLA -10T allele could be causal for the observed neurological manifestations. Future studies are needed to clarify whether affected patients benefit from GLA enzyme replacement therapy for end-organ damage prevention.
Figures
Similar articles
-
Alpha-Galactosidase A p.A143T, a non-Fabry disease-causing variant.Orphanet J Rare Dis. 2016 May 4;11(1):54. doi: 10.1186/s13023-016-0441-z. Orphanet J Rare Dis. 2016. PMID: 27142856 Free PMC article.
-
Frequency of Fabry disease in patients with small-fibre neuropathy of unknown aetiology: a pilot study.Eur J Neurol. 2011 Apr;18(4):631-6. doi: 10.1111/j.1468-1331.2010.03227.x. Epub 2010 Sep 23. Eur J Neurol. 2011. PMID: 20860754
-
An intronic haplotype in α galactosidase A is associated with reduced mRNA expression in males with cryptogenic stroke.Gene. 2014 Oct 10;549(2):275-9. doi: 10.1016/j.gene.2014.08.004. Epub 2014 Aug 4. Gene. 2014. PMID: 25101867
-
Neurological complications of Anderson-Fabry disease.Curr Pharm Des. 2013;19(33):6014-30. doi: 10.2174/13816128113199990387. Curr Pharm Des. 2013. PMID: 23448452 Review.
-
How relevant are cerebral white matter lesions in the D313Y variant of the α-galactosidase A gene? Neurological, cardiological, laboratory, and MRI data of 21 patients within a follow-up of 3 years.Neurol Sci. 2023 Apr;44(4):1375-1381. doi: 10.1007/s10072-022-06533-7. Epub 2022 Dec 2. Neurol Sci. 2023. PMID: 36456878 Review.
Cited by
-
Organ manifestations and long-term outcome of Fabry disease in patients with the GLA haplotype D313Y.BMJ Open. 2016 Apr 8;6(4):e010422. doi: 10.1136/bmjopen-2015-010422. BMJ Open. 2016. PMID: 27059467 Free PMC article.
-
Screening of Fabry disease in patients with end-stage renal disease of unknown etiology: the first Thailand study.J Biomed Res. 2016 Oct 17;31(1):17-24. doi: 10.7555/JBR.31.20160063. J Biomed Res. 2016. PMID: 28808181 Free PMC article.
-
Females with Fabry disease: an expert opinion on diagnosis, clinical management, current challenges and unmet needs.Front Cardiovasc Med. 2025 Mar 12;12:1536114. doi: 10.3389/fcvm.2025.1536114. eCollection 2025. Front Cardiovasc Med. 2025. PMID: 40144933 Free PMC article. Review.
-
Frequency of Fabry disease in a juvenile idiopathic arthritis cohort.Pediatr Rheumatol Online J. 2021 Jun 12;19(1):91. doi: 10.1186/s12969-021-00563-9. Pediatr Rheumatol Online J. 2021. PMID: 34118938 Free PMC article.
-
Alpha-Galactosidase A p.A143T, a non-Fabry disease-causing variant.Orphanet J Rare Dis. 2016 May 4;11(1):54. doi: 10.1186/s13023-016-0441-z. Orphanet J Rare Dis. 2016. PMID: 27142856 Free PMC article.
References
-
- Desnick RJ, Brady R, Barranger J, Collins AJ, Germain DP, Goldman M, Grabowski G, Packman S, Wilcox WR. Fabry disease, an under-recognized multisystemic disorder: expert recommendations for diagnosis, management, and enzyme replacement therapy. Ann Intern Med. 2003;138:338–346. doi: 10.7326/0003-4819-138-4-200302180-00014. - DOI - PubMed
-
- Mehta A, Ricci R, Widmer U, Dehout F, Garcia de Lorenzom A, Kampmann C, Linhart A, Sunder-Plassmann G, Ries M, Beck M. Fabry disease defined: baseline clinical manifestations of 366 patients in the Fabry Outcome Survey. Eur J Clin Invest. 2004;34:236–242. doi: 10.1111/j.1365-2362.2004.01309.x. - DOI - PubMed
-
- Oliveira JP, Ferreira S, Reguenga C, Carvalho F, Månsson JE. The g.1170C>T polymorphism of the 5′ untranslated region of the human alpha-galactosidase gene is associated with decreased enzyme expression–evidence from a family study. J Inherit Metab Dis. 2008;31:S405–S413. doi: 10.1007/s10545-008-0972-0. - DOI - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
