

Severe dystonia, cerebellar atrophy, and cardiomyopathy likely caused by a missense mutation in TOR1AIP1
- PMID: 25425325
- PMCID: PMC4302636
- DOI: 10.1186/s13023-014-0174-9
Severe dystonia, cerebellar atrophy, and cardiomyopathy likely caused by a missense mutation in TOR1AIP1
Abstract
Background: Dystonia, cerebellar atrophy, and cardiomyopathy constitute a rare association.
Methods: We used homozygosity mapping and whole exome sequencing to determine the mutation, western blot and immunolabelling on cultured fibroblasts to demonstrate the lower expression and the mislocalization of the protein.
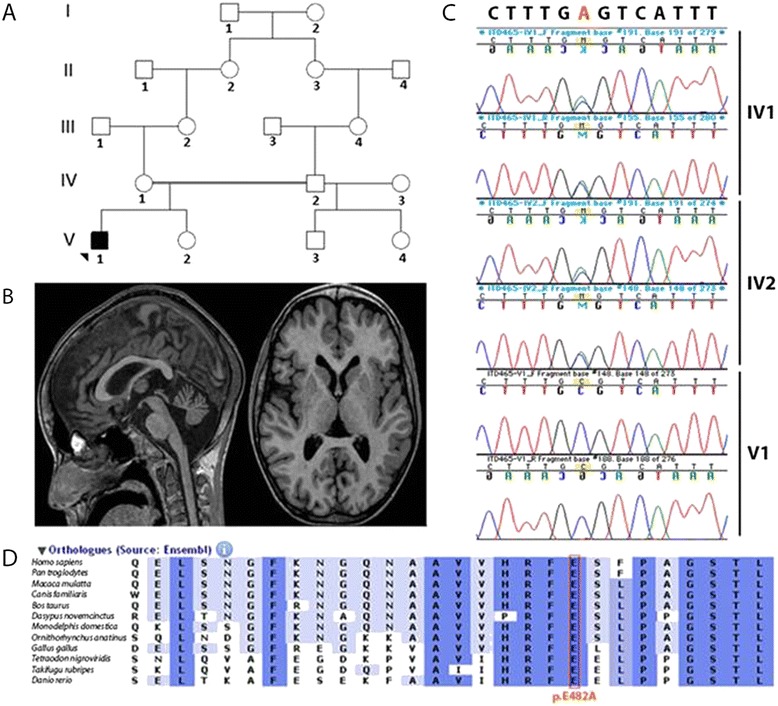
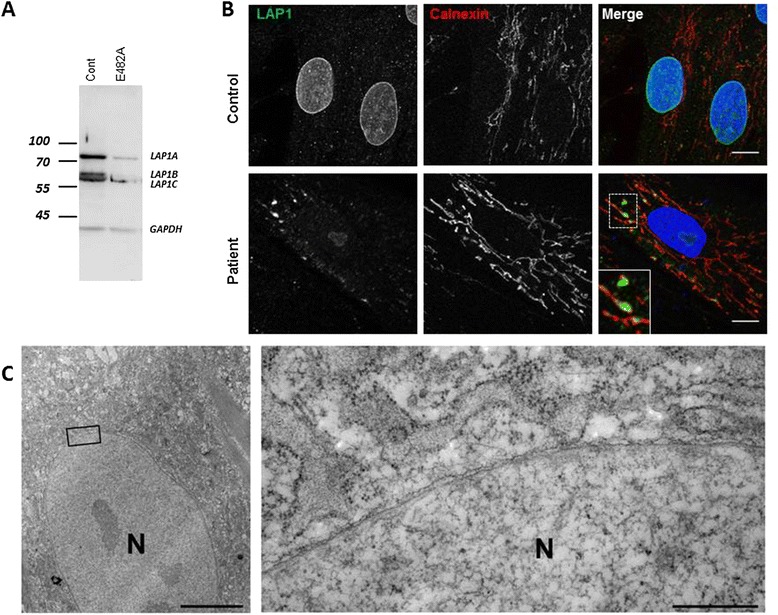
Results: We report on a boy born from consanguineous healthy parents, who presented at three years of age with rapidly progressing dystonia, progressive cerebellar atrophy, and dilated cardiomyopathy. We identified regions of homozygosity and performed whole exome sequencing that revealed a homozygous missense mutation in TOR1AIP1. The mutation, absent in controls, results in a change of a highly conserved glutamic acid to alanine. TOR1AIP1 encodes lamina-associated polypeptide 1 (LAP1), a transmembrane protein ubiquitously expressed in the inner nuclear membrane. LAP1 interacts with torsinA, the protein mutated in DYT1-dystonia. In vitro studies in fibroblasts of the patient revealed reduced expression of LAP1 and its mislocalization and aggregation in the endoplasmic reticulum as underlying pathogenic mechanisms.
Conclusions and relevance: The pathogenic role of TOR1AIP1 mutation is supported by a) the involvement of a highly conserved amino acid, b) the absence of the mutation in controls, c) the functional interaction of LAP1 with torsinA, and d) mislocalization of LAP1 in patient cells. Of note, cardiomyopathy has been reported in LAP1-null mice and in patients with the TOR1AIP1 nonsense mutation. Other cases will help delineate the clinical spectrum of LAP1-related mutations.
Figures
References
-
- Boukhris A, Schule R, Loureiro JL, Lourenço CM, Mundwiller E, Gonzalez MA, Charles P, Gauthier J, Rekik I, Acosta Lebrigio RF, Gaussen M, Speziani F, Ferbert A, Feki I, Caballero-Oteyza A, Dionne-Laporte A, Amri M, Noreau A, Forlani S, Cruz VT, Mochel F, Coutinho P, Dion P, Mhiri C, Schols L, Pouget J, Darios F, Rouleau GA, Marques W, Jr, Brice A, et al. Alteration of ganglioside biosynthesis responsible for complex hereditary spastic paraplegia. Am J Hum Genet. 2013;93(1):118–123. doi: 10.1016/j.ajhg.2013.05.006. - DOI - PMC - PubMed
-
- Cattin ME, Bertrand AT, Schlossarek S, Le Bihan MC, Skov Jensen S, Neuber C, Crocini C, Maron S, Lainé J, Mougenot N, Varnous S, Fromes Y, Hansen A, Eschenhagen T, Decostre V, Carrier L, Bonne G. Heterozygous LmnadelK32 mice develop dilated cardiomyopathy through a combined pathomechanism of haploinsufficiency and peptide toxicity. Hum Mol Genet. 2013;22(15):3152–3164. doi: 10.1093/hmg/ddt172. - DOI - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
