

Dissecting the clonal origins of childhood acute lymphoblastic leukemia by single-cell genomics
- PMID: 25425670
- PMCID: PMC4273416
- DOI: 10.1073/pnas.1420822111
Dissecting the clonal origins of childhood acute lymphoblastic leukemia by single-cell genomics
Abstract
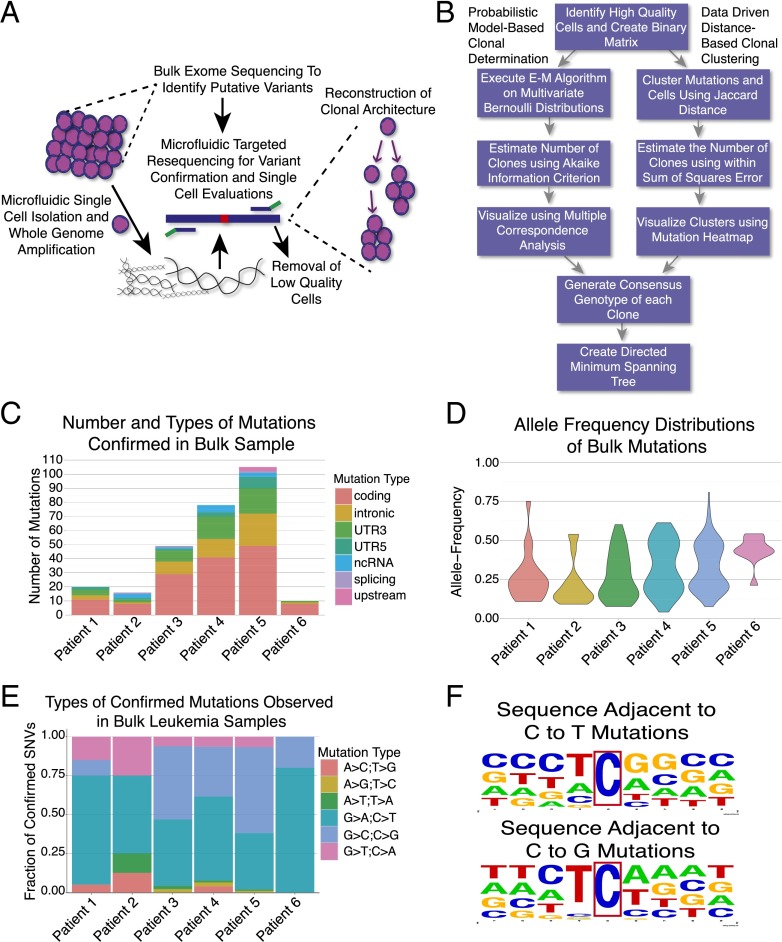
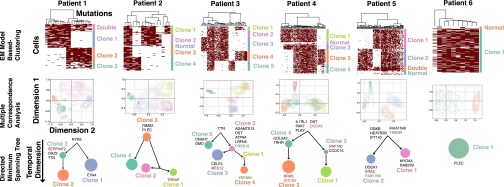
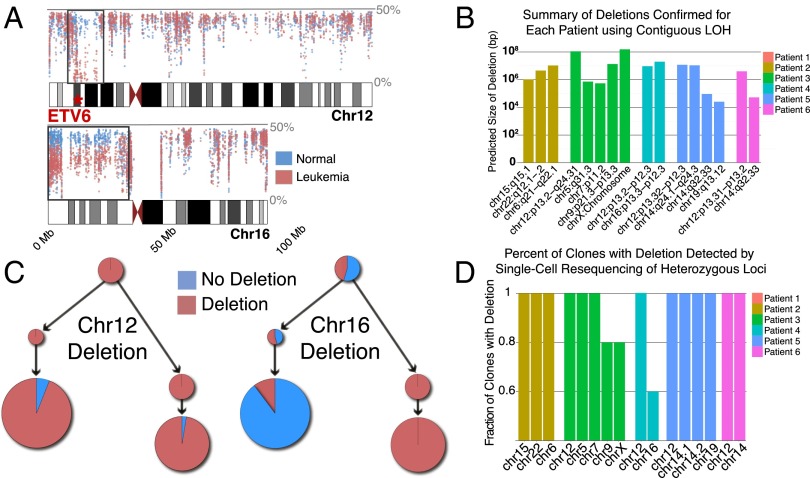
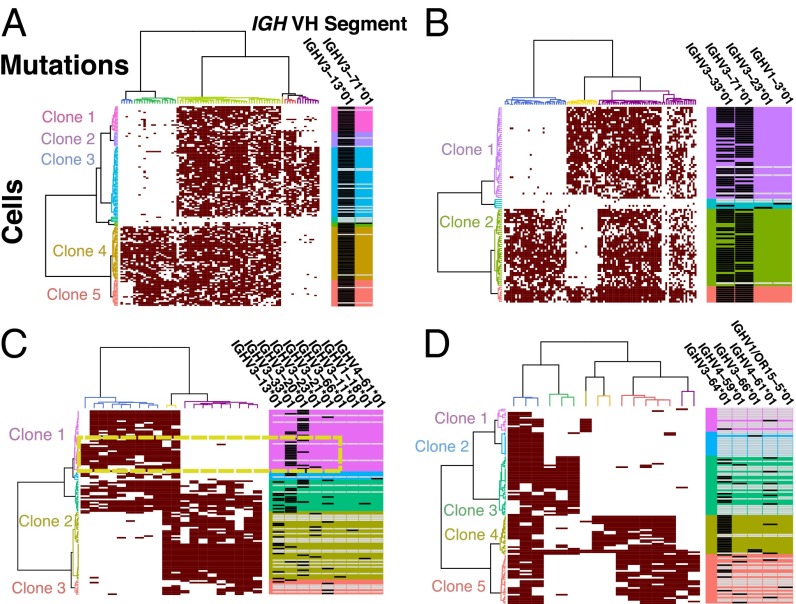
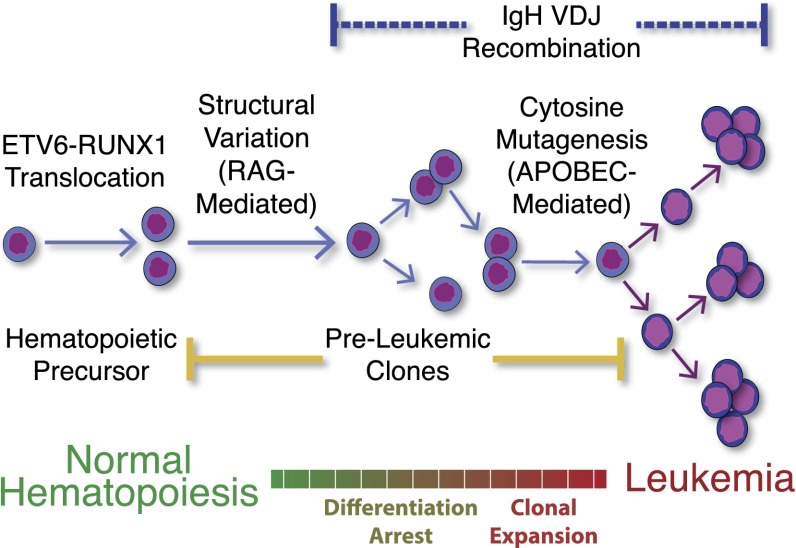
Many cancers have substantial genomic heterogeneity within a given tumor, and to fully understand that diversity requires the ability to perform single cell analysis. We performed targeted sequencing of a panel of single nucleotide variants (SNVs), deletions, and IgH sequences in 1,479 single tumor cells from six acute lymphoblastic leukemia (ALL) patients. By accurately segregating groups of cooccurring mutations into distinct clonal populations, we identified codominant clones in the majority of patients. Evaluation of intraclonal mutation patterns identified clone-specific punctuated cytosine mutagenesis events, showed that most structural variants are acquired before SNVs, determined that KRAS mutations occur late in disease development but are not sufficient for clonal dominance, and identified clones within the same patient that are arrested at varied stages in B-cell development. Taken together, these data order the sequence of genetic events that underlie childhood ALL and provide a framework for understanding the development of the disease at single-cell resolution.
Keywords: acute lymphoblastic leukemia; clonal evolution; cytosine mutagenesis; intratumor heterogeneity; single-cell genomics.
Conflict of interest statement
Conflict of interest statement: S.R.Q. is a founder of and consultant for Fluidigm.
Figures
References
Publication types
MeSH terms
Associated data
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
