

Versatility of the complement system in neuroinflammation, neurodegeneration and brain homeostasis
- PMID: 25426028
- PMCID: PMC4224073
- DOI: 10.3389/fncel.2014.00380
Versatility of the complement system in neuroinflammation, neurodegeneration and brain homeostasis
Erratum in
-
Corrigendum: Versatility of the complement system in neuroinflammation, neurodegeneration, and brain homeostasis.Front Cell Neurosci. 2015 Jul 7;9:263. doi: 10.3389/fncel.2015.00263. eCollection 2015. Front Cell Neurosci. 2015. PMID: 26217187 Free PMC article.
Abstract
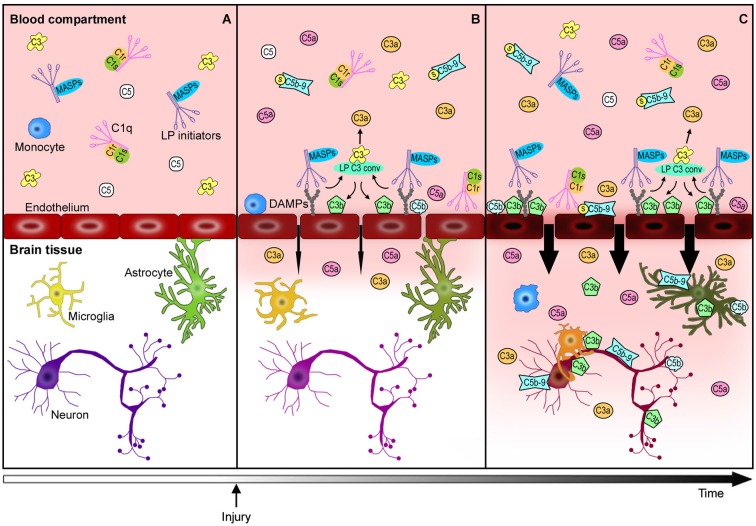
The immune response after brain injury is highly complex and involves both local and systemic events at the cellular and molecular level. It is associated to a dramatic over-activation of enzyme systems, the expression of proinflammatory genes and the activation/recruitment of immune cells. The complement system represents a powerful component of the innate immunity and is highly involved in the inflammatory response. Complement components are synthesized predominantly by the liver and circulate in the bloodstream primed for activation. Moreover, brain cells can produce complement proteins and receptors. After acute brain injury, the rapid and uncontrolled activation of the complement leads to massive release of inflammatory anaphylatoxins, recruitment of cells to the injury site, phagocytosis and induction of blood brain barrier (BBB) damage. Brain endothelial cells are particularly susceptible to complement-mediated effects, since they are exposed to both circulating and locally synthesized complement proteins. Conversely, during neurodegenerative disorders, complement factors play distinct roles depending on the stage and degree of neuropathology. In addition to the deleterious role of the complement, increasing evidence suggest that it may also play a role in normal nervous system development (wiring the brain) and adulthood (either maintaining brain homeostasis or supporting regeneration after brain injury). This article represents a compendium of the current knowledge on the complement role in the brain, prompting a novel view that complement activation can result in either protective or detrimental effects in brain conditions that depend exquisitely on the nature, the timing and the degree of the stimuli that induce its activation. A deeper understanding of the acute, subacute and chronic consequences of complement activation is needed and may lead to new therapeutic strategies, including the ability of targeting selective step in the complement cascade.
Keywords: Alzheimer’s disease; brain homeostasis; complement system; endothelium; stroke; therapeutic targets; traumatic brain injury.
Figures

References
-
- Arumugam T. V., Tang S.-C., Lathia J. D., Cheng A., Mughal M. R., Chigurupati S., et al. . (2007). Intravenous immunoglobulin (IVIG) protects the brain against experimental stroke by preventing complement-mediated neuronal cell death. Proc. Natl. Acad. Sci. U S A 104, 14104–14109. 10.1073/pnas.0700506104 - DOI - PMC - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources