

BreaKmer: detection of structural variation in targeted massively parallel sequencing data using kmers
- PMID: 25428359
- PMCID: PMC4330340
- DOI: 10.1093/nar/gku1211
BreaKmer: detection of structural variation in targeted massively parallel sequencing data using kmers
Abstract
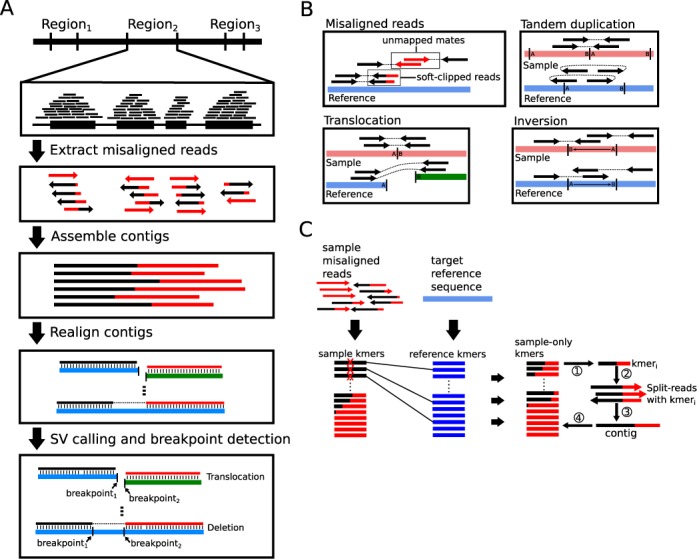
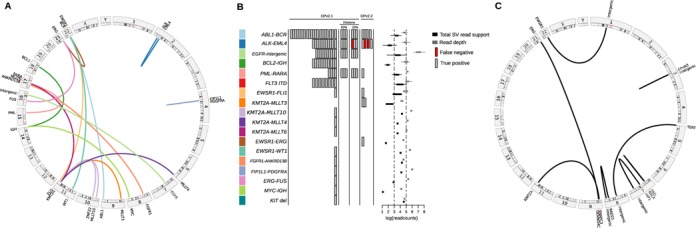
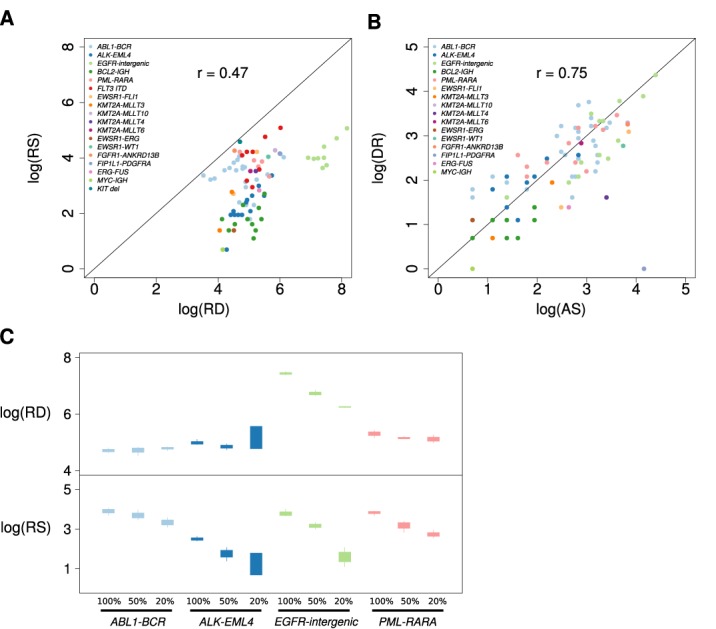
Genomic structural variation (SV), a common hallmark of cancer, has important predictive and therapeutic implications. However, accurately detecting SV using high-throughput sequencing data remains challenging, especially for 'targeted' resequencing efforts. This is critically important in the clinical setting where targeted resequencing is frequently being applied to rapidly assess clinically actionable mutations in tumor biopsies in a cost-effective manner. We present BreaKmer, a novel approach that uses a 'kmer' strategy to assemble misaligned sequence reads for predicting insertions, deletions, inversions, tandem duplications and translocations at base-pair resolution in targeted resequencing data. Variants are predicted by realigning an assembled consensus sequence created from sequence reads that were abnormally aligned to the reference genome. Using targeted resequencing data from tumor specimens with orthogonally validated SV, non-tumor samples and whole-genome sequencing data, BreaKmer had a 97.4% overall sensitivity for known events and predicted 17 positively validated, novel variants. Relative to four publically available algorithms, BreaKmer detected SV with increased sensitivity and limited calls in non-tumor samples, key features for variant analysis of tumor specimens in both the clinical and research settings.
© The Author(s) 2014. Published by Oxford University Press on behalf of Nucleic Acids Research.
Figures
References
-
- Mitelman F., Johansson B., Mertens F. The impact of translocations and gene fusions on cancer causation. Nat. Rev. Cancer. 2007;7:233–245. - PubMed
-
- Rowley J.D. Chromosome translocations: dangerous liaisons revisited. Nat. Rev. Cancer. 2001;1:245–250. - PubMed
-
- Rowley J.D. Chromosomal translocations: revisited yet again. Blood. 2008;112:2183–2189. - PubMed
Publication types
MeSH terms
Substances
Associated data
LinkOut - more resources
Full Text Sources
Other Literature Sources
