

Congenital myopathies: Natural history of a large pediatric cohort
- PMID: 25428687
- PMCID: PMC4336094
- DOI: 10.1212/WNL.0000000000001110
Congenital myopathies: Natural history of a large pediatric cohort
Abstract
Objective: To assess the natural history of congenital myopathies (CMs) due to different genotypes.
Methods: Retrospective cross-sectional study based on case-note review of 125 patients affected by CM, followed at a single pediatric neuromuscular center, between 1984 and 2012.
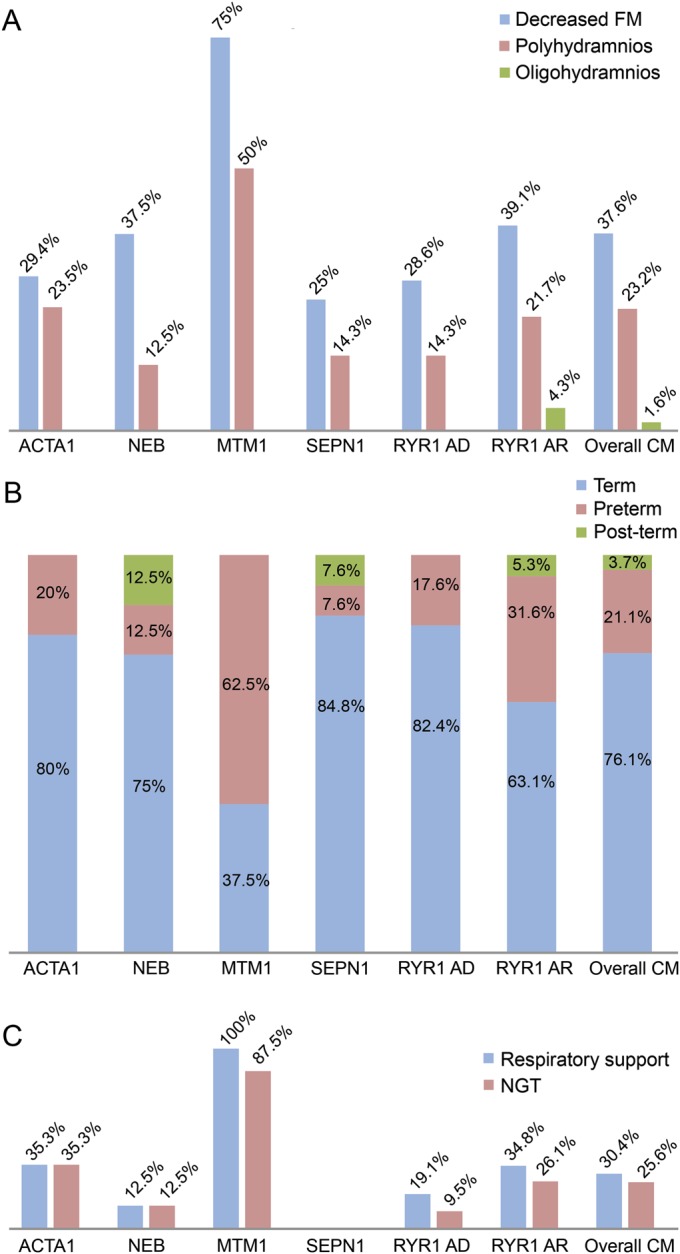
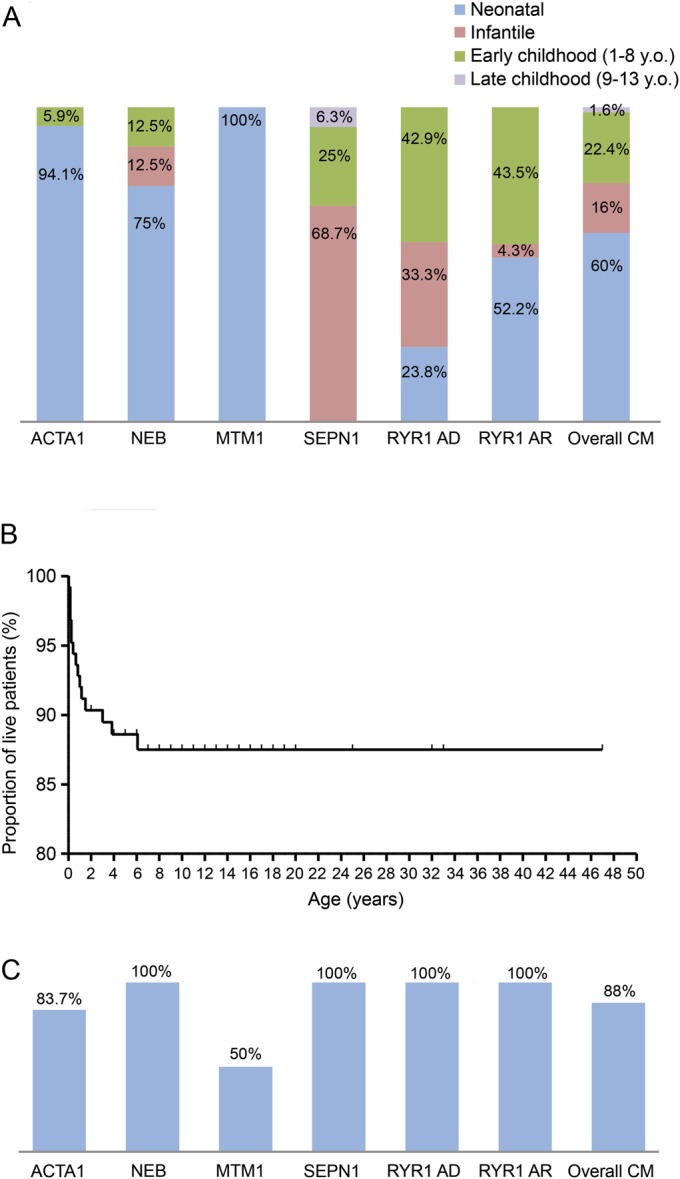
Results: Genetic characterization was achieved in 99 of 125 cases (79.2%), with RYR1 most frequently implicated (44/125). Neonatal/infantile onset was observed in 76%. At birth, 30.4% required respiratory support, and 25.2% nasogastric feeding. Twelve percent died, mainly within the first year, associated with mutations in ACTA1, MTM1, or KLHL40. All RYR1-mutated cases survived and did not require long-term ventilator support including those with severe neonatal onset; however, recessive cases were more likely to require gastrostomy insertion (p = 0.0028) compared with dominant cases. Independent ambulation was achieved in 74.1% of all patients; 62.9% were late walkers. Among ambulant patients, 9% eventually became wheelchair-dependent. Scoliosis of variable severity was reported in 40%, with 1/3 of (both ambulant and nonambulant) patients requiring surgery. Bulbar involvement was present in 46.4% and required gastrostomy placement in 28.8% (at a mean age of 2.7 years). Respiratory impairment of variable severity was a feature in 64.1%; approximately half of these patients required nocturnal noninvasive ventilation due to respiratory failure (at a mean age of 8.5 years).
Conclusions: We describe the long-term outcome of a large cohort of patients with CMs. While overall course is stable, we demonstrate a wide clinical spectrum with motor deterioration in a subset of cases. Severity in the neonatal/infantile period is critical for survival, with clear genotype-phenotype correlations that may inform future counseling.
© 2014 American Academy of Neurology.
Figures


Comment in
-
Congenital myopathies: Rebuilding the natural history, one gene at a time.Neurology. 2015 Jan 6;84(1):15-6. doi: 10.1212/WNL.0000000000001117. Epub 2014 Nov 26. Neurology. 2015. PMID: 25428690 No abstract available.
References
-
- Sewry C, Jimenez-Mallebrera C, Muntoni F. Congenital myopathies. Curr Opin Neurol 2008;21:569–575. - PubMed
-
- Laing N. Congenital myopathies. Curr Opin Neurol 2007;20:583–589. - PubMed
-
- Scoto M, Cirak S, Mein R, et al. SEPN1-related myopathies: clinical course in a large cohort of patients. Neurology 2011;76:2073–2078. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources