

CMT subtypes and disease burden in patients enrolled in the Inherited Neuropathies Consortium natural history study: a cross-sectional analysis
- PMID: 25430934
- PMCID: PMC4516002
- DOI: 10.1136/jnnp-2014-308826
CMT subtypes and disease burden in patients enrolled in the Inherited Neuropathies Consortium natural history study: a cross-sectional analysis
Abstract
Background: The international Inherited Neuropathy Consortium (INC) was created with the goal of obtaining much needed natural history data for patients with Charcot-Marie-Tooth (CMT) disease. We analysed clinical and genetic data from patients in the INC to determine the distribution of CMT subtypes and the clinical impairment associated with them.
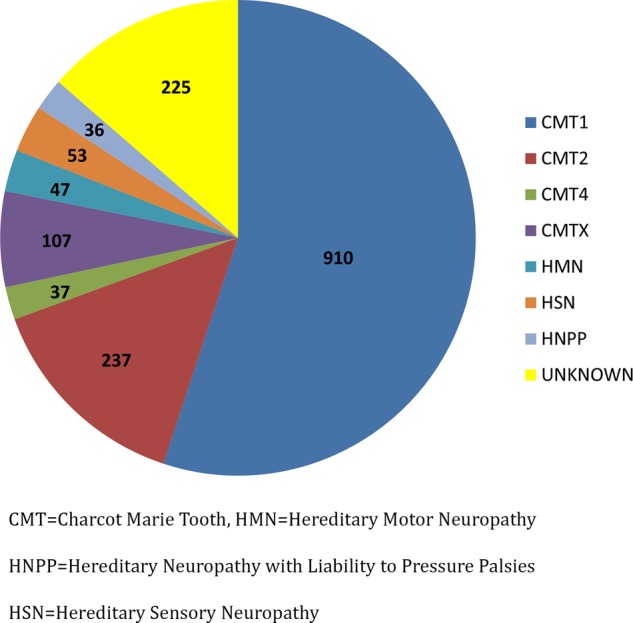
Methods: We analysed data from 1652 patients evaluated at 13 INC centres. The distribution of CMT subtypes and pathogenic genetic mutations were determined. The disease burden of all the mutations was assessed by the CMT Neuropathy Score (CMTNS) and CMT Examination Score (CMTES).
Results: 997 of the 1652 patients (60.4%) received a genetic diagnosis. The most common CMT subtypes were CMT1A/PMP22 duplication, CMT1X/GJB1 mutation, CMT2A/MFN2 mutation, CMT1B/MPZ mutation, and hereditary neuropathy with liability to pressure palsy/PMP22 deletion. These five subtypes of CMT accounted for 89.2% of all genetically confirmed mutations. Mean CMTNS for some but not all subtypes were similar to those previously reported.
Conclusions: Our findings confirm that large numbers of patients with a representative variety of CMT subtypes have been enrolled and that the frequency of achieving a molecular diagnosis and distribution of the CMT subtypes reflects those previously reported. Measures of severity are similar, though not identical, to results from smaller series. This study confirms that it is possible to assess patients in a uniform way between international centres, which is critical for the planned natural history study and future clinical trials. These data will provide a representative baseline for longitudinal studies of CMT.
Clinical trial registration: ID number NCT01193075.
Keywords: GENETICS; NEUROGENETICS; NEUROPATHY.
Published by the BMJ Publishing Group Limited. For permission to use (where not already granted under a licence) please go to http://group.bmj.com/group/rights-licensing/permissions.
Figures
References
-
- Skre H. Genetic and clinical aspects of Charcot-Marie-Tooth's disease. Clin Genet 1974;6:98–118. - PubMed
-
- Rossor AM, Polke JM, Houlden H, et al. . Clinical implications of genetic advances in Charcot-Marie-Tooth disease. Nat Rev Neurol 2013;9:562–71. - PubMed
-
- Shy ME, Blake J, Krajewski K, et al. . Reliability and validity of the CMT neuropathy score as a measure of disability. Neurology 2005;64:1209–14. - PubMed
-
- Shy ME, Chen L, Swan ER, et al. . Neuropathy progression in Charcot-Marie-Tooth disease type 1A. Neurology 2008;70:378–83. - PubMed
Publication types
MeSH terms
Substances
Associated data
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases