

Elimination of cap structures generated by mRNA decay involves the new scavenger mRNA decapping enzyme Aph1/FHIT together with DcpS
- PMID: 25432955
- PMCID: PMC4288156
- DOI: 10.1093/nar/gku1251
Elimination of cap structures generated by mRNA decay involves the new scavenger mRNA decapping enzyme Aph1/FHIT together with DcpS
Abstract
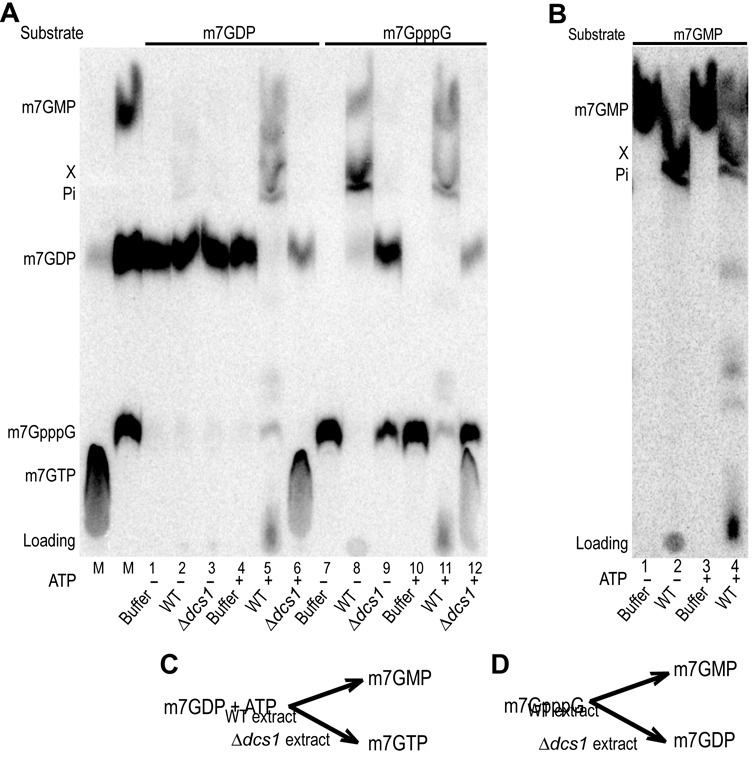
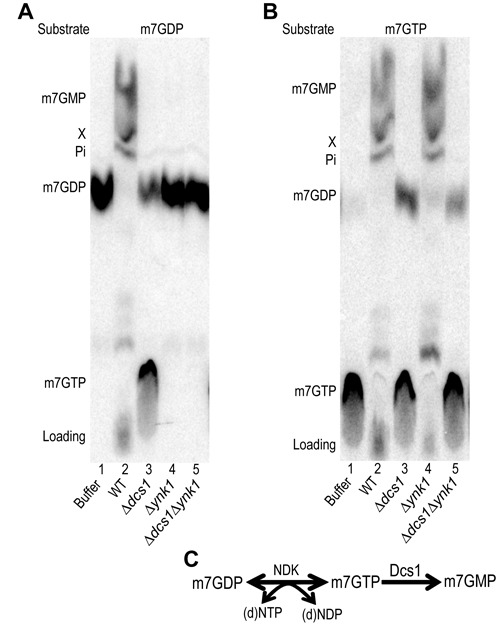
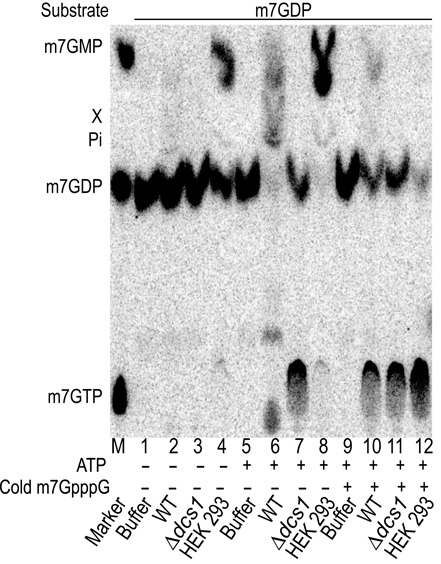
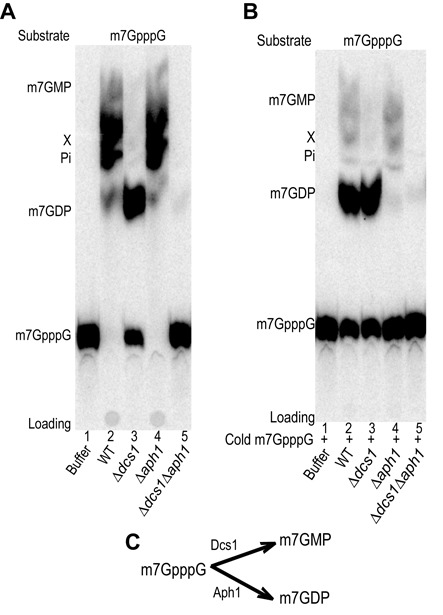
Eukaryotic 5' mRNA cap structures participate to the post-transcriptional control of gene expression before being released by the two main mRNA decay pathways. In the 3'-5' pathway, the exosome generates free cap dinucleotides (m7GpppN) or capped oligoribonucleotides that are hydrolyzed by the Scavenger Decapping Enzyme (DcpS) forming m7GMP. In the 5'-3' pathway, the decapping enzyme Dcp2 generates m7GDP. We investigated the fate of m7GDP and m7GpppN produced by RNA decay in extracts and cells. This defined a pathway involving DcpS, NTPs and the nucleoside diphosphate kinase for m7GDP elimination. Interestingly, we identified and characterized in vitro and in vivo a new scavenger decapping enzyme involved in m7GpppN degradation. We show that activities mediating cap elimination identified in yeast are essentially conserved in human. Their alteration may contribute to pathologies, possibly through the interference of cap (di)nucleotide with cellular function.
© The Author(s) 2014. Published by Oxford University Press on behalf of Nucleic Acids Research.
Figures
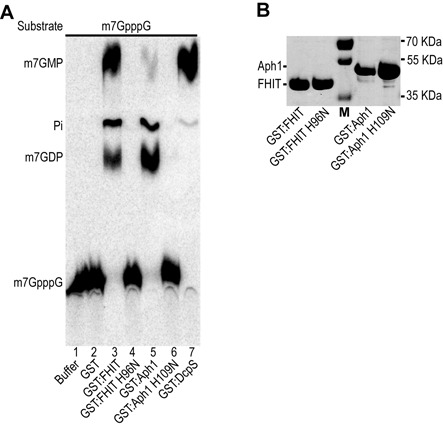
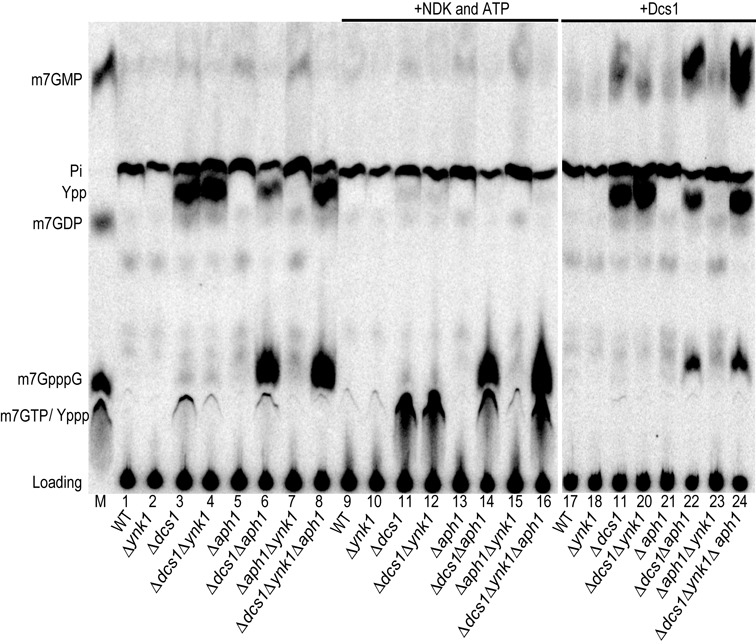
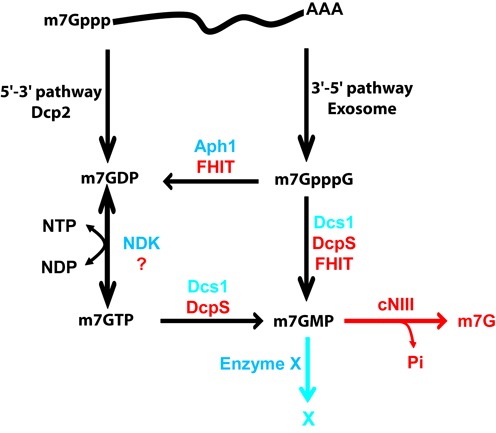
References
-
- Fischer U., Englbrecht C., Chari A. Biogenesis of spliceosomal small nuclear ribonucleoproteins. Wiley Interdiscip. Rev. RNA. 2011;2:718–731. - PubMed
-
- Blumenthal T. Trans-splicing and polycistronic transcription in Caenorhabditis elegans. Trends Genet. 1995;11:132–136. - PubMed
-
- Bangs J.D., Crain P.F., Hashizume T., McCloskey J.A., Boothroyd J.C. Mass spectrometry of mRNA cap 4 from trypanosomatids reveals two novel nucleosides. J. Biol. Chem. 1992;267:9805–9815. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
