

Leveraging transcription factors to speed cellobiose fermentation by Saccharomyces cerevisiae
- PMID: 25435910
- PMCID: PMC4243952
- DOI: 10.1186/s13068-014-0126-6
Leveraging transcription factors to speed cellobiose fermentation by Saccharomyces cerevisiae
Abstract
Background: Saccharomyces cerevisiae, a key organism used for the manufacture of renewable fuels and chemicals, has been engineered to utilize non-native sugars derived from plant cell walls, such as cellobiose and xylose. However, the rates and efficiencies of these non-native sugar fermentations pale in comparison with those of glucose. Systems biology methods, used to understand biological networks, hold promise for rational microbial strain development in metabolic engineering. Here, we present a systematic strategy for optimizing non-native sugar fermentation by recombinant S. cerevisiae, using cellobiose as a model.
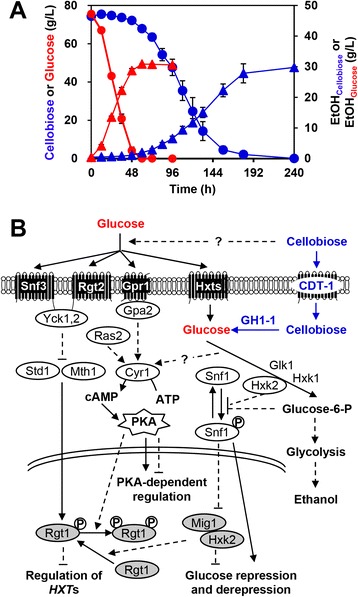
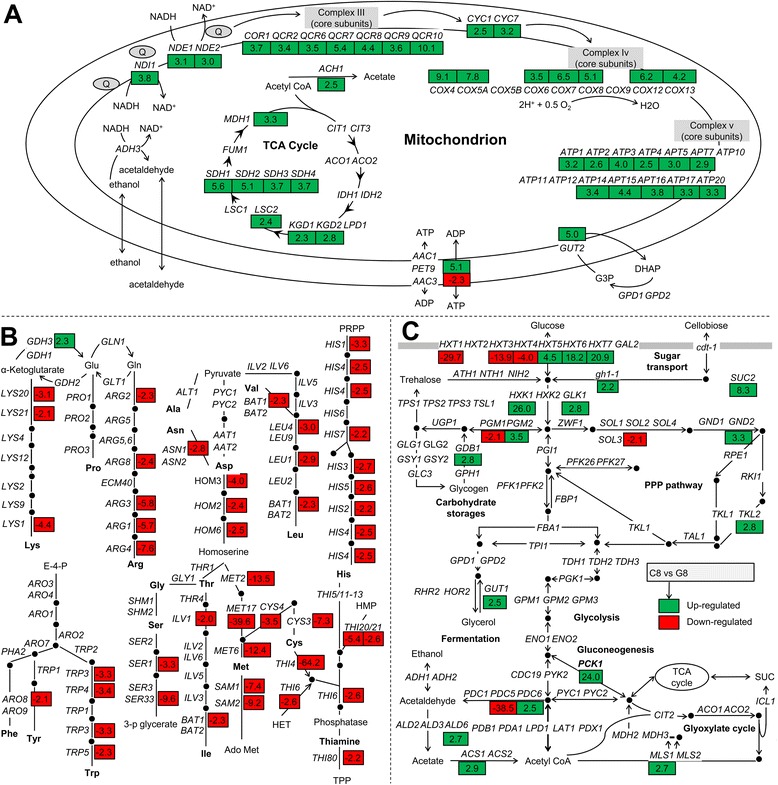
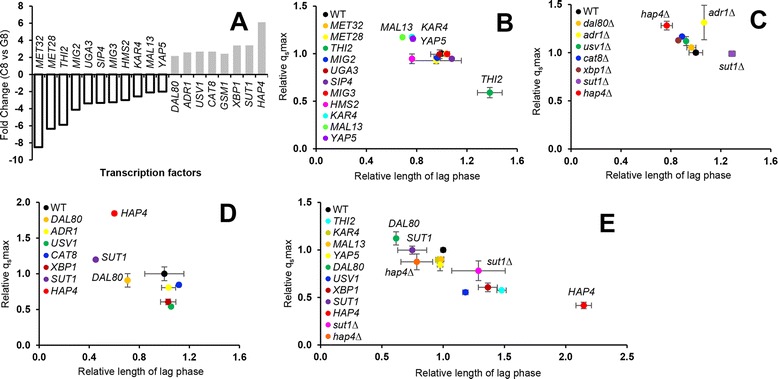
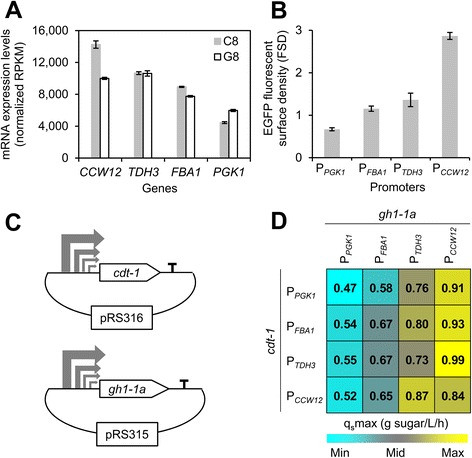
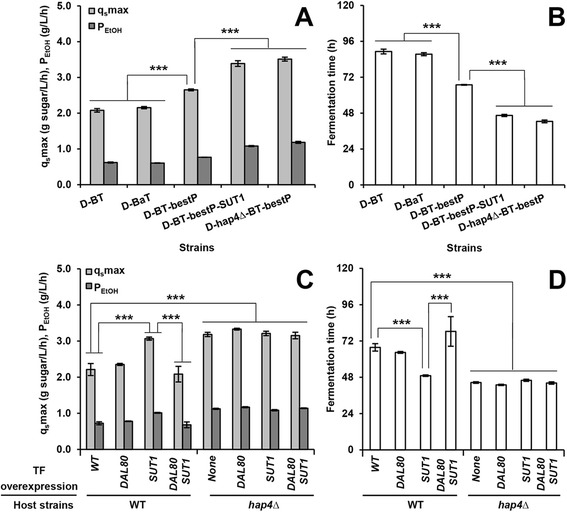
Results: Differences in gene expression between cellobiose and glucose metabolism revealed by RNA deep sequencing indicated that cellobiose metabolism induces mitochondrial activation and reduces amino acid biosynthesis under fermentation conditions. Furthermore, glucose-sensing and signaling pathways and their target genes, including the cAMP-dependent protein kinase A pathway controlling the majority of glucose-induced changes, the Snf3-Rgt2-Rgt1 pathway regulating hexose transport, and the Snf1-Mig1 glucose repression pathway, were at most only partially activated under cellobiose conditions. To separate correlations from causative effects, the expression levels of 19 transcription factors perturbed under cellobiose conditions were modulated, and the three strongest promoters under cellobiose conditions were applied to fine-tune expression of the heterologous cellobiose-utilizing pathway. Of the changes in these 19 transcription factors, only overexpression of SUT1 or deletion of HAP4 consistently improved cellobiose fermentation. SUT1 overexpression and HAP4 deletion were not synergistic, suggesting that SUT1 and HAP4 may regulate overlapping genes important for improved cellobiose fermentation. Transcription factor modulation coupled with rational tuning of the cellobiose consumption pathway significantly improved cellobiose fermentation.
Conclusions: We used systems-level input to reveal the regulatory mechanisms underlying suboptimal metabolism of the non-glucose sugar cellobiose. By identifying key transcription factors that cause suboptimal cellobiose fermentation in engineered S. cerevisiae, and by fine-tuning the expression of a heterologous cellobiose consumption pathway, we were able to greatly improve cellobiose fermentation by engineered S. cerevisiae. Our results demonstrate a powerful strategy for applying systems biology methods to rapidly identify metabolic engineering targets and overcome bottlenecks in performance of engineered strains.
Keywords: Biofuels; Cellobiose; Glycolysis; Metabolic engineering; Systems biology; Transcription factor.
Figures
Similar articles
-
Gene Amplification on Demand Accelerates Cellobiose Utilization in Engineered Saccharomyces cerevisiae.Appl Environ Microbiol. 2016 May 31;82(12):3631-3639. doi: 10.1128/AEM.00410-16. Print 2016 Jun 15. Appl Environ Microbiol. 2016. PMID: 27084006 Free PMC article.
-
[Effect of MIG1 and SNF1 deletion on simultaneous utilization of glucose and xylose by Saccharomyces cerevisiae].Sheng Wu Gong Cheng Xue Bao. 2018 Jan 25;34(1):54-67. doi: 10.13345/j.cjb.170098. Sheng Wu Gong Cheng Xue Bao. 2018. PMID: 29380571 Chinese.
-
Improving Xylose Utilization of Saccharomyces cerevisiae by Expressing the MIG1 Mutant from the Self-Flocculating Yeast SPSC01.Protein Pept Lett. 2018;25(2):202-207. doi: 10.2174/0929866525666180122142609. Protein Pept Lett. 2018. PMID: 29359658
-
Strain engineering of Saccharomyces cerevisiae for enhanced xylose metabolism.Biotechnol Adv. 2013 Nov;31(6):851-61. doi: 10.1016/j.biotechadv.2013.03.004. Epub 2013 Mar 21. Biotechnol Adv. 2013. PMID: 23524005 Review.
-
Production of fuels and chemicals from xylose by engineered Saccharomyces cerevisiae: a review and perspective.Microb Cell Fact. 2017 May 11;16(1):82. doi: 10.1186/s12934-017-0694-9. Microb Cell Fact. 2017. PMID: 28494761 Free PMC article. Review.
Cited by
-
A semi-synthetic regulon enables rapid growth of yeast on xylose.Nat Commun. 2018 Mar 26;9(1):1233. doi: 10.1038/s41467-018-03645-7. Nat Commun. 2018. PMID: 29581426 Free PMC article.
-
QTL analysis reveals genomic variants linked to high-temperature fermentation performance in the industrial yeast.Biotechnol Biofuels. 2019 Mar 19;12:59. doi: 10.1186/s13068-019-1398-7. eCollection 2019. Biotechnol Biofuels. 2019. PMID: 30923567 Free PMC article.
-
Engineering transcription factors to improve tolerance against alkane biofuels in Saccharomyces cerevisiae.Biotechnol Biofuels. 2015 Dec 30;8:231. doi: 10.1186/s13068-015-0411-z. eCollection 2015. Biotechnol Biofuels. 2015. PMID: 26719765 Free PMC article.
-
Expanding xylose metabolism in yeast for plant cell wall conversion to biofuels.Elife. 2015 Feb 3;4:e05896. doi: 10.7554/eLife.05896. Elife. 2015. PMID: 25647728 Free PMC article.
-
Mechanisms underlying lactic acid tolerance and its influence on lactic acid production in Saccharomyces cerevisiae.Microb Cell. 2021 Apr 14;8(6):111-130. doi: 10.15698/mic2021.06.751. Microb Cell. 2021. PMID: 34055965 Free PMC article. Review.
References
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
