

Reversible blockade of complex I or inhibition of PKCβ reduces activation and mitochondria translocation of p66Shc to preserve cardiac function after ischemia
- PMID: 25436907
- PMCID: PMC4250075
- DOI: 10.1371/journal.pone.0113534
Reversible blockade of complex I or inhibition of PKCβ reduces activation and mitochondria translocation of p66Shc to preserve cardiac function after ischemia
Abstract
Aim: Excess mitochondrial reactive oxygen species (mROS) play a vital role in cardiac ischemia reperfusion (IR) injury. P66Shc, a splice variant of the ShcA adaptor protein family, enhances mROS production by oxidizing reduced cytochrome c to yield H2O2. Ablation of p66Shc protects against IR injury, but it is unknown if and when p66Shc is activated during cardiac ischemia and/or reperfusion and if attenuating complex I electron transfer or deactivating PKCβ alters p66Shc activation during IR is associated with cardioprotection.
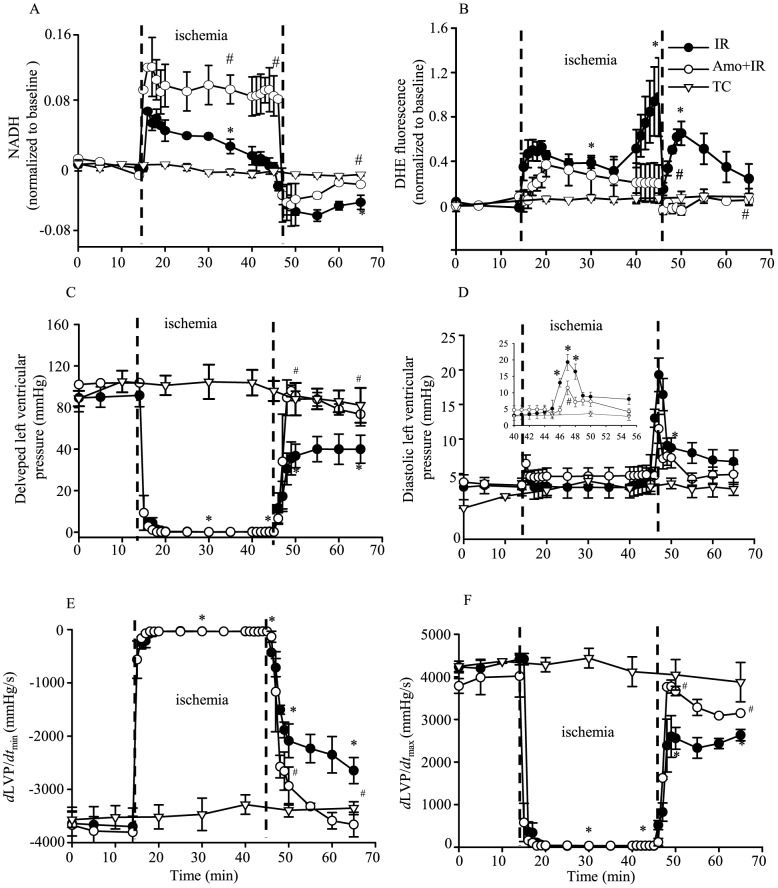
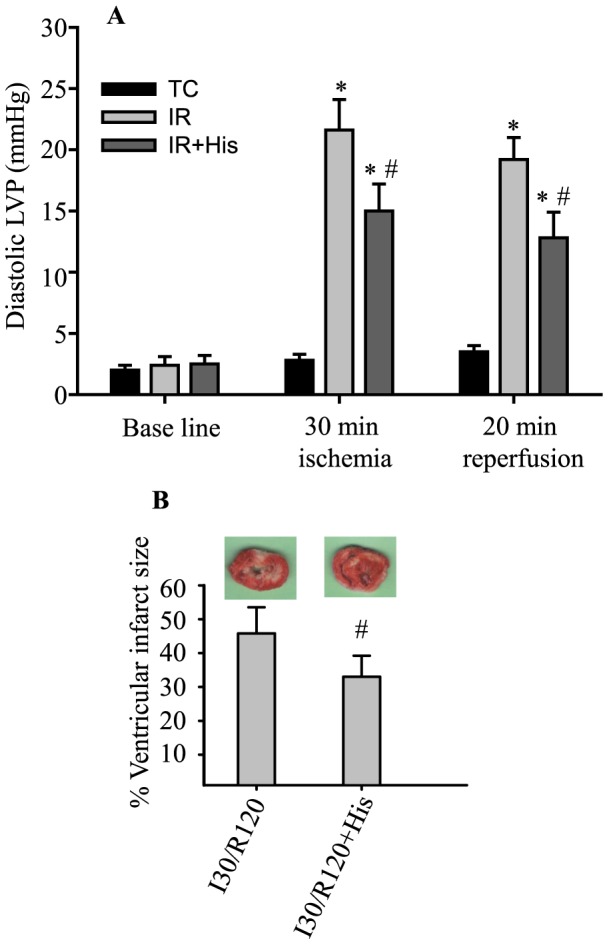
Methods: Isolated guinea pig hearts were perfused and subjected to increasing periods of ischemia and reperfusion with or without amobarbital, a complex I blocker, or hispidin, a PKCβ inhibitor. Phosphorylation of p66Shc at serine 36 and levels of p66Shc in mitochondria and cytosol were measured. Cardiac functional variables and redox states were monitored online before, during and after ischemia. Infarct size was assessed in some hearts after 120 min reperfusion.
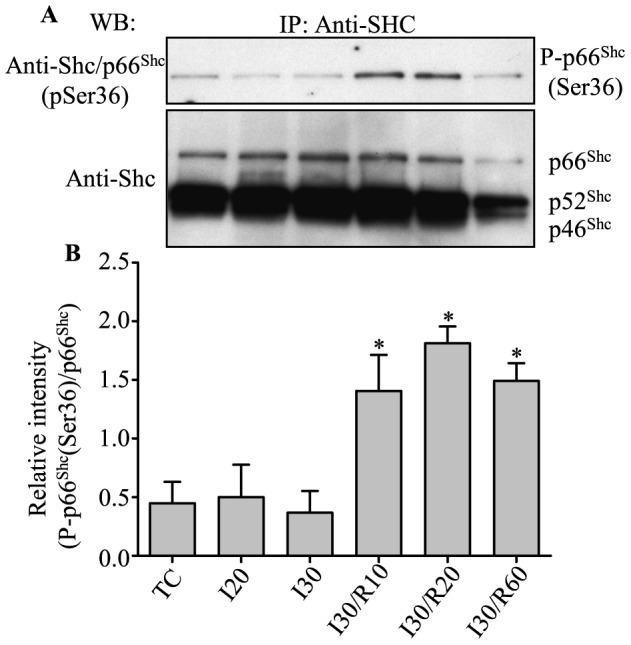
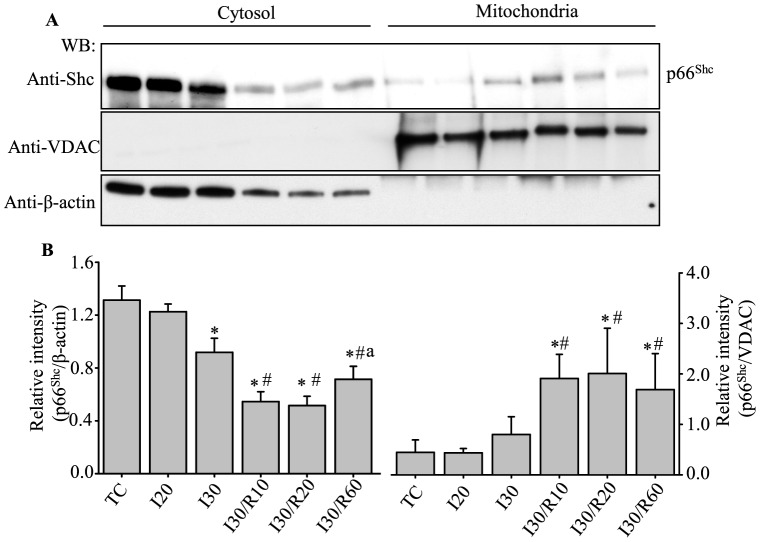
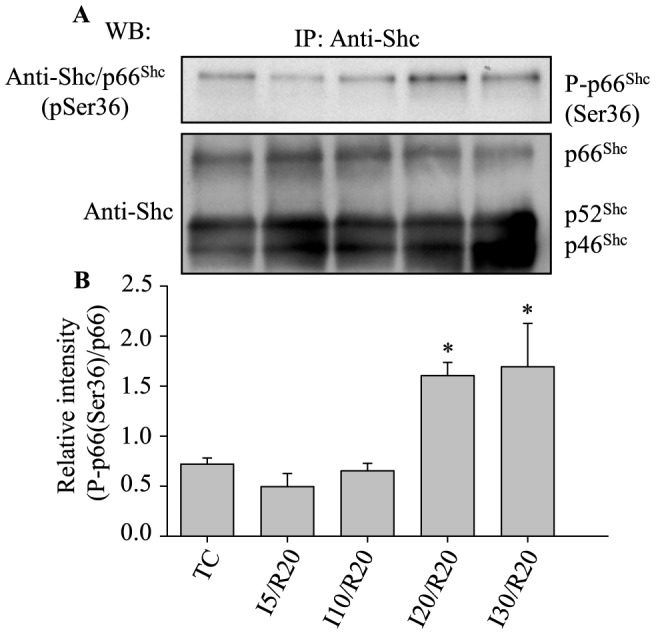
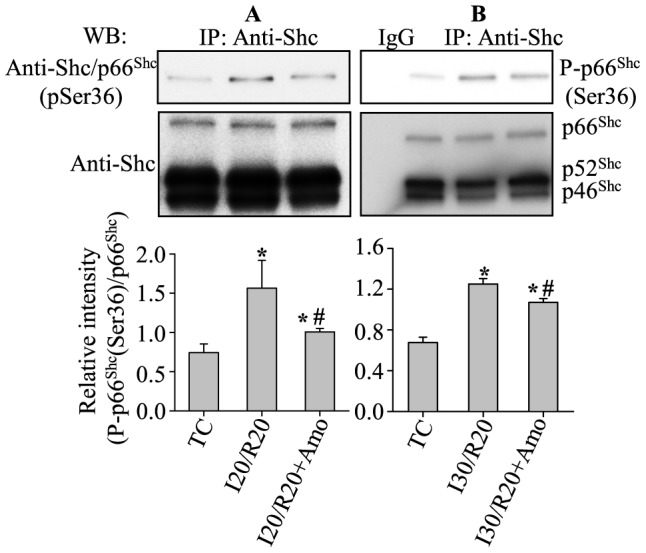
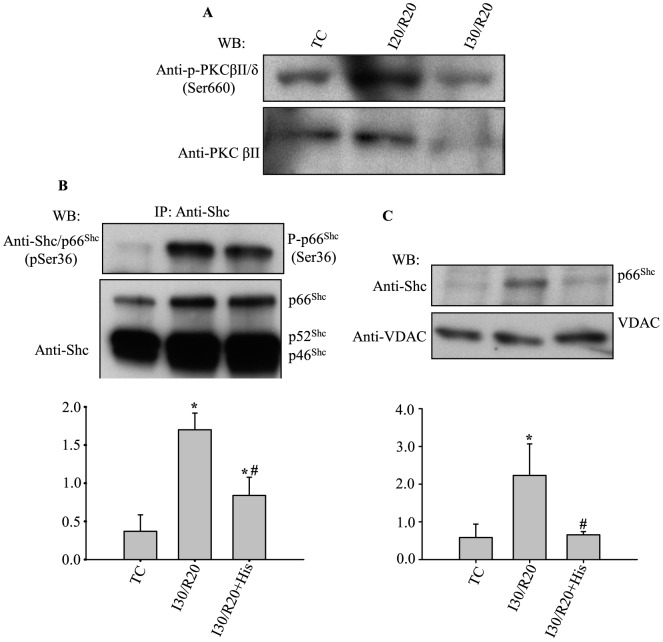
Results: Phosphorylation of p66Shc and its translocation into mitochondria increased during reperfusion after 20 and 30 min ischemia, but not during ischemia only, or during 5 or 10 min ischemia followed by 20 min reperfusion. Correspondingly, cytosolic p66Shc levels decreased during these ischemia and reperfusion periods. Amobarbital or hispidin reduced phosphorylation of p66Shc and its mitochondrial translocation induced by 30 min ischemia and 20 min reperfusion. Decreased phosphorylation of p66Shc by amobarbital or hispidin led to better functional recovery and less infarction during reperfusion.
Conclusion: Our results show that IR activates p66Shc and that reversible blockade of electron transfer from complex I, or inhibition of PKCβ activation, decreases p66Shc activation and translocation and reduces IR damage. These observations support a novel potential therapeutic intervention against cardiac IR injury.
Conflict of interest statement
Figures
Similar articles
-
p66Shc in Cardiovascular Pathology.Cells. 2022 Jun 6;11(11):1855. doi: 10.3390/cells11111855. Cells. 2022. PMID: 35681549 Free PMC article. Review.
-
Inhibited mitochondrial respiration by amobarbital during cardiac ischaemia improves redox state and reduces matrix Ca2+ overload and ROS release.Cardiovasc Res. 2008 Jan 15;77(2):406-15. doi: 10.1016/j.cardiores.2007.08.008. Cardiovasc Res. 2008. PMID: 17900548
-
Blockade of PKCβ protects against remote organ injury induced by intestinal ischemia and reperfusion via a p66shc-mediated mitochondrial apoptotic pathway.Apoptosis. 2014 Sep;19(9):1342-53. doi: 10.1007/s10495-014-1008-x. Apoptosis. 2014. PMID: 24930012
-
Blockade of electron transport before cardiac ischemia with the reversible inhibitor amobarbital protects rat heart mitochondria.J Pharmacol Exp Ther. 2006 Jan;316(1):200-7. doi: 10.1124/jpet.105.091702. Epub 2005 Sep 20. J Pharmacol Exp Ther. 2006. PMID: 16174799
-
P66shc and its role in ischemic cardiovascular diseases.Basic Res Cardiol. 2019 Jun 4;114(4):29. doi: 10.1007/s00395-019-0738-x. Basic Res Cardiol. 2019. PMID: 31165272 Review.
Cited by
-
Mitochondria-Associated Endoplasmic Reticulum Membranes in the Pathogenesis of Type 2 Diabetes Mellitus.Front Cell Dev Biol. 2020 Oct 20;8:571554. doi: 10.3389/fcell.2020.571554. eCollection 2020. Front Cell Dev Biol. 2020. PMID: 33195204 Free PMC article. Review.
-
Metformin attenuates ER stress-induced mitochondrial dysfunction.Transl Res. 2017 Dec;190:40-50. doi: 10.1016/j.trsl.2017.09.003. Epub 2017 Sep 28. Transl Res. 2017. PMID: 29040818 Free PMC article.
-
Mitochondrial VDAC1: A Key Gatekeeper as Potential Therapeutic Target.Front Physiol. 2017 Jun 30;8:460. doi: 10.3389/fphys.2017.00460. eCollection 2017. Front Physiol. 2017. PMID: 28713289 Free PMC article. Review.
-
p66Shc in Cardiovascular Pathology.Cells. 2022 Jun 6;11(11):1855. doi: 10.3390/cells11111855. Cells. 2022. PMID: 35681549 Free PMC article. Review.
-
Maslinic acid alleviates ischemia/reperfusion-induced inflammation by downregulation of NFκB-mediated adhesion molecule expression.Sci Rep. 2019 Apr 16;9(1):6119. doi: 10.1038/s41598-019-42465-7. Sci Rep. 2019. PMID: 30992483 Free PMC article.
References
-
- Chen Q, Vazquez EJ, Moghaddas S, Hoppel CL, Lesnefsky EJ (2003) Production of reactive oxygen species by mitochondria: Central role of complex III. J Biol Chem 278:36027–36031. - PubMed
-
- St-Pierre J, Buckingham JA, Roebuck SJ, Brand MD (2002) Topology of superoxide production from different sites in the mitochondrial electron transport chain. J Biol Chem 277:44784–44790. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
