

PRIMUS: rapid reconstruction of pedigrees from genome-wide estimates of identity by descent
- PMID: 25439724
- PMCID: PMC4225580
- DOI: 10.1016/j.ajhg.2014.10.005
PRIMUS: rapid reconstruction of pedigrees from genome-wide estimates of identity by descent
Abstract
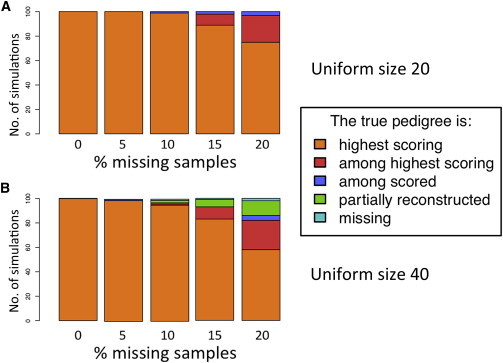
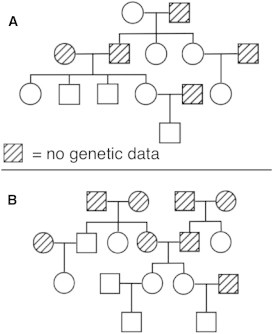
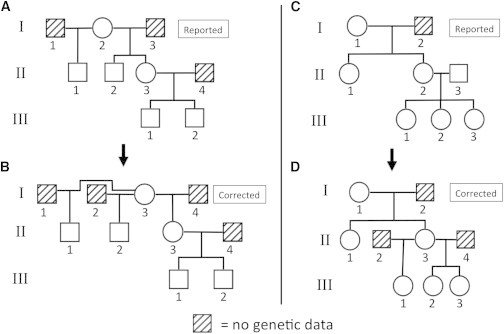
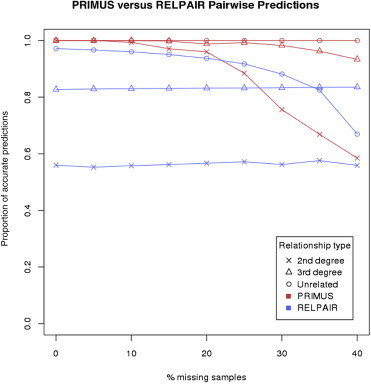
Understanding and correctly utilizing relatedness among samples is essential for genetic analysis; however, managing sample records and pedigrees can often be error prone and incomplete. Data sets ascertained by random sampling often harbor cryptic relatedness that can be leveraged in genetic analyses for maximizing power. We have developed a method that uses genome-wide estimates of pairwise identity by descent to identify families and quickly reconstruct and score all possible pedigrees that fit the genetic data by using up to third-degree relatives, and we have included it in the software package PRIMUS (Pedigree Reconstruction and Identification of the Maximally Unrelated Set). Here, we validate its performance on simulated, clinical, and HapMap pedigrees. Among these samples, we demonstrate that PRIMUS can verify reported pedigree structures and identify cryptic relationships. Finally, we show that PRIMUS reconstructed pedigrees, all of which were previously unknown, for 203 families from a cohort collected in Starr County, TX (1,890 samples).
Copyright © 2014 The American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Figures

References
-
- Santorico S.A., Edwards K.L. Challenges of linkage analysis in the era of whole-genome sequencing. Genet. Epidemiol. 2014;38(Suppl 1):S92–S96. - PubMed
-
- Ott J., Kamatani Y., Lathrop M. Family-based designs for genome-wide association studies. Nat. Rev. Genet. 2011;12:465–474. - PubMed
-
- Below J.E., Earl D.L., Shively K.M., McMillin M.J., Smith J.D., Turner E.H., Stephan M.J., Al-Gazali L.I., Hertecant J.L., Chitayat D., University of Washington Center for Mendelian Genomics Whole-genome analysis reveals that mutations in inositol polyphosphate phosphatase-like 1 cause opsismodysplasia. Am. J. Hum. Genet. 2013;92:137–143. - PMC - PubMed
Publication types
MeSH terms
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
