

Mutations in SNX14 cause a distinctive autosomal-recessive cerebellar ataxia and intellectual disability syndrome
- PMID: 25439728
- PMCID: PMC4225633
- DOI: 10.1016/j.ajhg.2014.10.007
Mutations in SNX14 cause a distinctive autosomal-recessive cerebellar ataxia and intellectual disability syndrome
Erratum in
- Am J Hum Genet. 2015 Jun 4;96(6):1008-9
Abstract
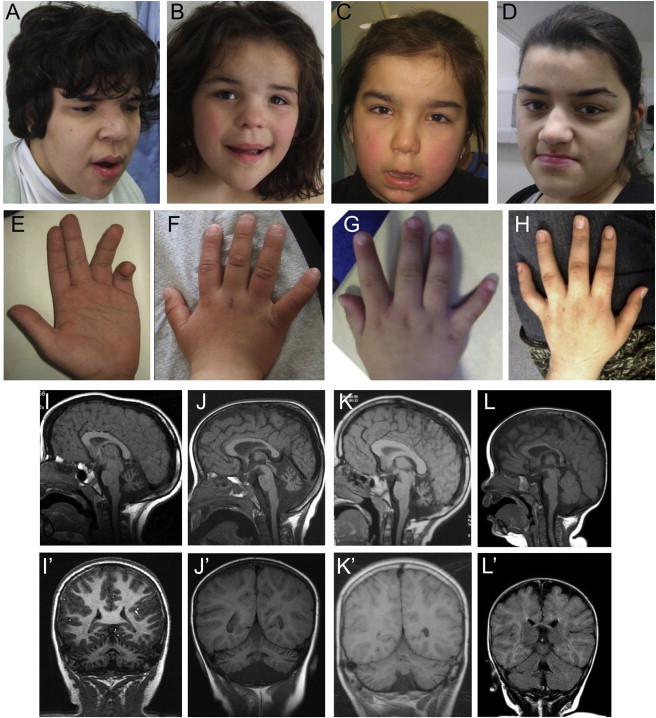
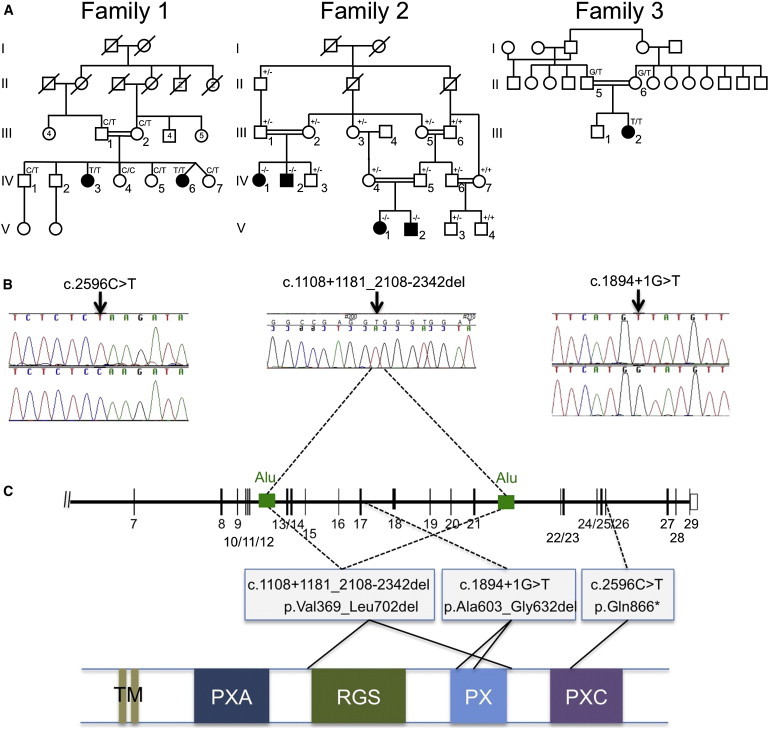
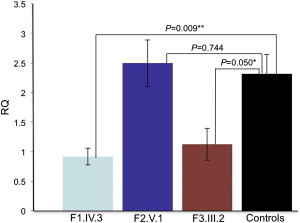
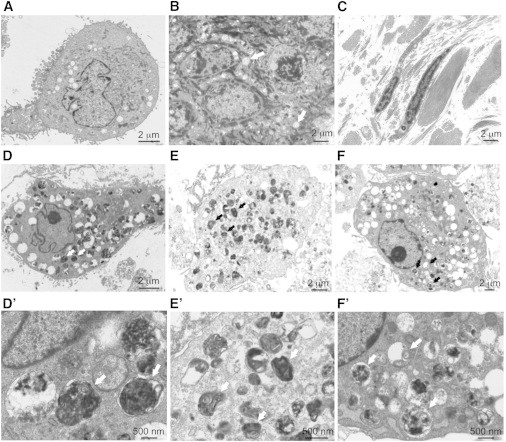
Intellectual disability and cerebellar atrophy occur together in a large number of genetic conditions and are frequently associated with microcephaly and/or epilepsy. Here we report the identification of causal mutations in Sorting Nexin 14 (SNX14) found in seven affected individuals from three unrelated consanguineous families who presented with recessively inherited moderate-severe intellectual disability, cerebellar ataxia, early-onset cerebellar atrophy, sensorineural hearing loss, and the distinctive association of progressively coarsening facial features, relative macrocephaly, and the absence of seizures. We used homozygosity mapping and whole-exome sequencing to identify a homozygous nonsense mutation and an in-frame multiexon deletion in two families. A homozygous splice site mutation was identified by Sanger sequencing of SNX14 in a third family, selected purely by phenotypic similarity. This discovery confirms that these characteristic features represent a distinct and recognizable syndrome. SNX14 encodes a cellular protein containing Phox (PX) and regulator of G protein signaling (RGS) domains. Weighted gene coexpression network analysis predicts that SNX14 is highly coexpressed with genes involved in cellular protein metabolism and vesicle-mediated transport. All three mutations either directly affected the PX domain or diminished SNX14 levels, implicating a loss of normal cellular function. This manifested as increased cytoplasmic vacuolation as observed in cultured fibroblasts. Our findings indicate an essential role for SNX14 in neural development and function, particularly in development and maturation of the cerebellum.
Copyright © 2014 The Authors. Published by Elsevier Inc. All rights reserved.
Figures
References
-
- Ezgu F., Krejci P., Li S., de Sousa C., Graham J.M., Jr., Hansmann I., He W., Porpora K., Wand D., Wertelecki W. Phenotype-genotype correlations in patients with Marinesco-Sjögren syndrome. Clin. Genet. 2014;86:74–84. - PubMed
-
- Strehle E.M. Sialic acid storage disease and related disorders. Genet. Test. 2003;7:113–121. - PubMed
-
- Waterham H.R., Ebberink M.S. Genetics and molecular basis of human peroxisome biogenesis disorders. Biochim. Biophys. Acta. 2012;1822:1430–1441. - PubMed
-
- Matthijs G., Schollen E., Pardon E., Veiga-Da-Cunha M., Jaeken J., Cassiman J.J., Van Schaftingen E. Mutations in PMM2, a phosphomannomutase gene on chromosome 16p13, in carbohydrate-deficient glycoprotein type I syndrome (Jaeken syndrome) Nat. Genet. 1997;16:88–92. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
