

Mutations in CKAP2L, the human homolog of the mouse Radmis gene, cause Filippi syndrome
- PMID: 25439729
- PMCID: PMC4225581
- DOI: 10.1016/j.ajhg.2014.10.008
Mutations in CKAP2L, the human homolog of the mouse Radmis gene, cause Filippi syndrome
Abstract
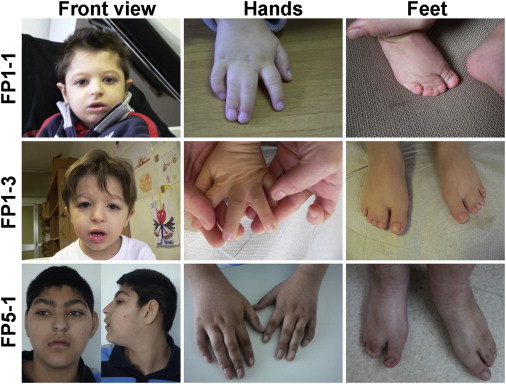
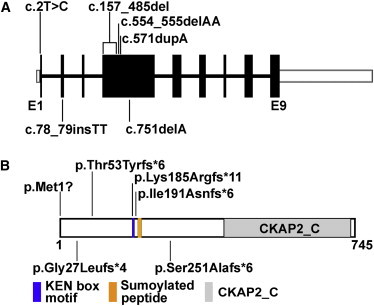
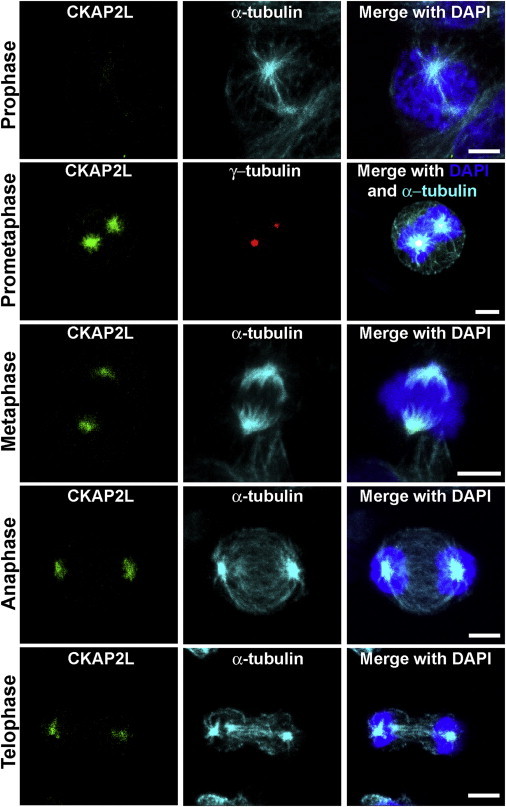
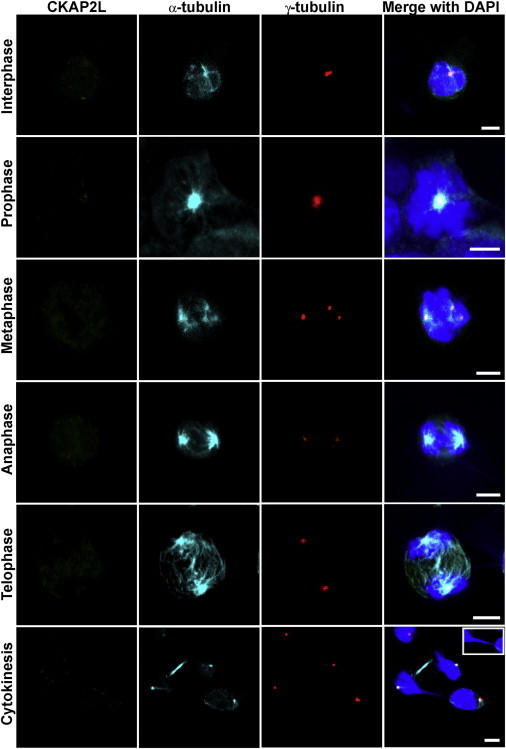
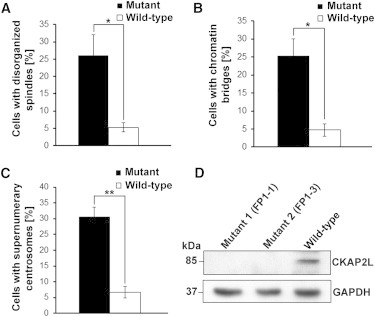
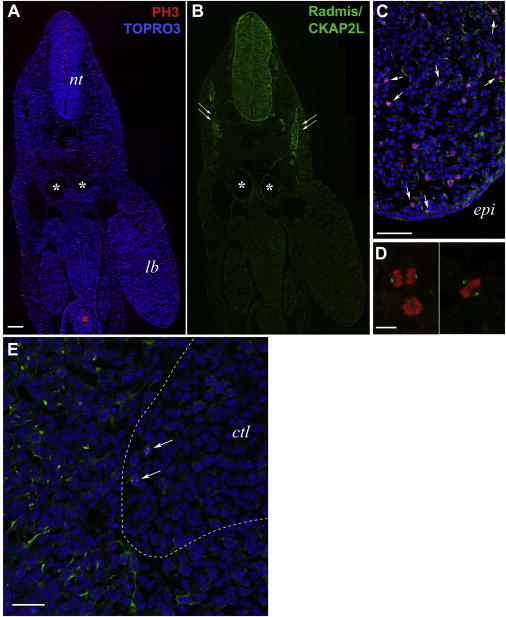
Filippi syndrome is a rare, presumably autosomal-recessive disorder characterized by microcephaly, pre- and postnatal growth failure, syndactyly, and distinctive facial features, including a broad nasal bridge and underdeveloped alae nasi. Some affected individuals have intellectual disability, seizures, undescended testicles in males, and teeth and hair abnormalities. We performed homozygosity mapping and whole-exome sequencing in a Sardinian family with two affected children and identified a homozygous frameshift mutation, c.571dupA (p.Ile191Asnfs(∗)6), in CKAP2L, encoding the protein cytoskeleton-associated protein 2-like (CKAP2L). The function of this protein was unknown until it was rediscovered in mice as Radmis (radial fiber and mitotic spindle) and shown to play a pivotal role in cell division of neural progenitors. Sanger sequencing of CKAP2L in a further eight unrelated individuals with clinical features consistent with Filippi syndrome revealed biallelic mutations in four subjects. In contrast to wild-type lymphoblastoid cell lines (LCLs), dividing LCLs established from the individuals homozygous for the c.571dupA mutation did not show CKAP2L at the spindle poles. Furthermore, in cells from the affected individuals, we observed an increase in the number of disorganized spindle microtubules owing to multipolar configurations and defects in chromosome segregation. The observed cellular phenotypes are in keeping with data from in vitro and in vivo knockdown studies performed in human cells and mice, respectively. Our findings show that loss-of-function mutations in CKAP2L are a major cause of Filippi syndrome.
Copyright © 2014 The American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Figures
References
-
- Meinecke P. Short stature, microcephaly, characteristic face, syndactyly and mental retardation: the Filippi syndrome. Report on a second family. Genet. Couns. 1993;4:147–151. - PubMed
-
- Filippi G. Unusual facial appearance, microcephaly, growth and mental retardation, and syndactyly. A new syndrome? Am. J. Med. Genet. 1985;22:821–824. - PubMed
-
- Orrico A., Hayek G. An additional case of craniodigital syndrome: variable expression of the Filippi syndrome? Clin. Genet. 1997;52:177–179. - PubMed
-
- Walpole I.R., Parry T., Goldblatt J. Expanding the phenotype of Filippi syndrome: a report of three cases. Clin. Dysmorphol. 1999;8:235–240. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
