

Transcriptome structure variability in Saccharomyces cerevisiae strains determined with a newly developed assembly software
- PMID: 25441755
- PMCID: PMC4302112
- DOI: 10.1186/1471-2164-15-1045
Transcriptome structure variability in Saccharomyces cerevisiae strains determined with a newly developed assembly software
Abstract
Background: RNA-seq studies have an important role for both large-scale analysis of gene expression and for transcriptome reconstruction. However, the lack of software specifically developed for the analysis of the transcriptome structure in lower eukaryotes, has so far limited the comparative studies among different species and strains.
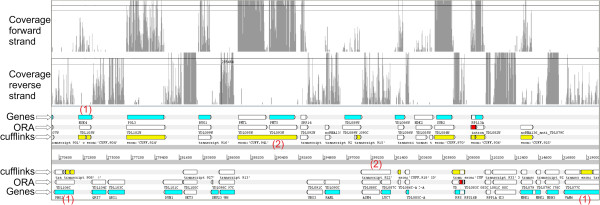
Results: In order to fill this gap, an innovative software called ORA (Overlapped Reads Assembler) was developed. This software allows a simple and reliable analysis of the transcriptome structure in organisms with a low number of introns. It can also determine the size and the position of the untranslated regions (UTR) and of polycistronic transcripts. As a case study, we analyzed the transcriptional landscape of six S. cerevisiae strains in two different key steps of the fermentation process. This comparative analysis revealed differences in the UTR regions of transcripts. By extending the transcriptome analysis to yeast species belonging to the Saccharomyces genus, it was possible to examine the conservation level of unknown non-coding RNAs and their putative functional role.
Conclusions: By comparing the results obtained using ORA with previous studies and with the transcriptome structure determined with other software, it was proven that ORA has a remarkable reliability. The results obtained from the training set made it possible to detect the presence of transcripts with variable UTRs between S. cerevisiae strains. Finally, we propose a regulatory role for some non-coding transcripts conserved within the Saccharomyces genus and localized in the antisense strand to genes involved in meiosis and cell wall biosynthesis.
Figures





References
-
- Sung HM, Wang TY, Wang D, Huang YS, Wu JP, Tsai HK, Tzeng J, Huang CJ, Lee YC, Yang P, Hsu J, Chang T, Cho CY, Weng LC, Lee TC, Chang TH, Li WH, Shih MC. Roles of trans and cis variation in yeast intraspecies evolution of gene expression. Mol Biol Evol. 2009;26(11):2533–2538. doi: 10.1093/molbev/msp171. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
