

The Hitchhiker's guide to Hi-C analysis: practical guidelines
- PMID: 25448293
- PMCID: PMC4347522
- DOI: 10.1016/j.ymeth.2014.10.031
The Hitchhiker's guide to Hi-C analysis: practical guidelines
Abstract
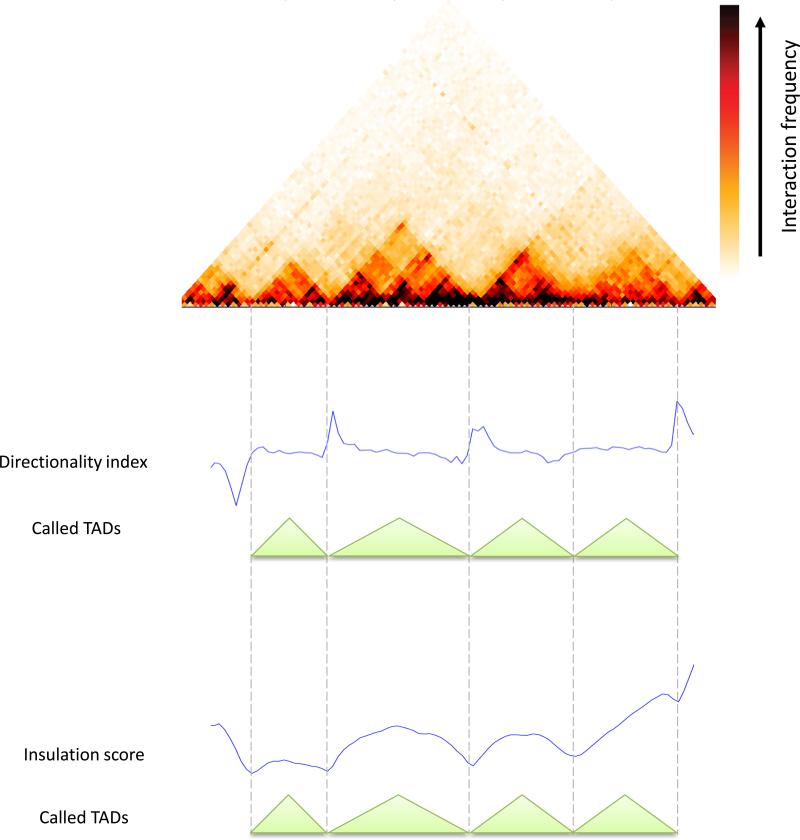
Over the last decade, development and application of a set of molecular genomic approaches based on the chromosome conformation capture method (3C), combined with increasingly powerful imaging approaches, have enabled high resolution and genome-wide analysis of the spatial organization of chromosomes. The aim of this paper is to provide guidelines for analyzing and interpreting data obtained with genome-wide 3C methods such as Hi-C and 3C-seq that rely on deep sequencing to detect and quantify pairwise chromatin interactions.
Keywords: Bioinformatics; Chromatin structure; Chromosome conformation capture; Deep sequencing; Hi-C.
Copyright © 2014 Elsevier Inc. All rights reserved.
Figures
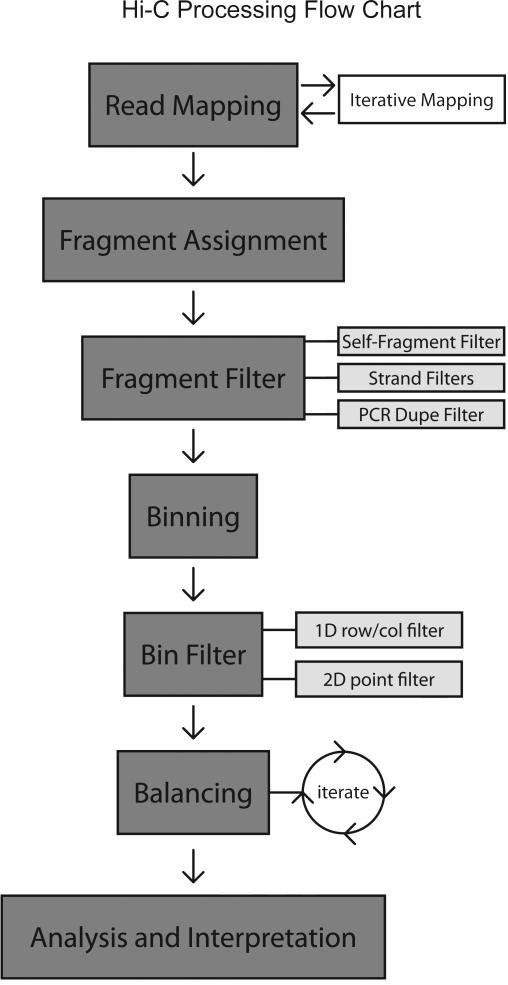
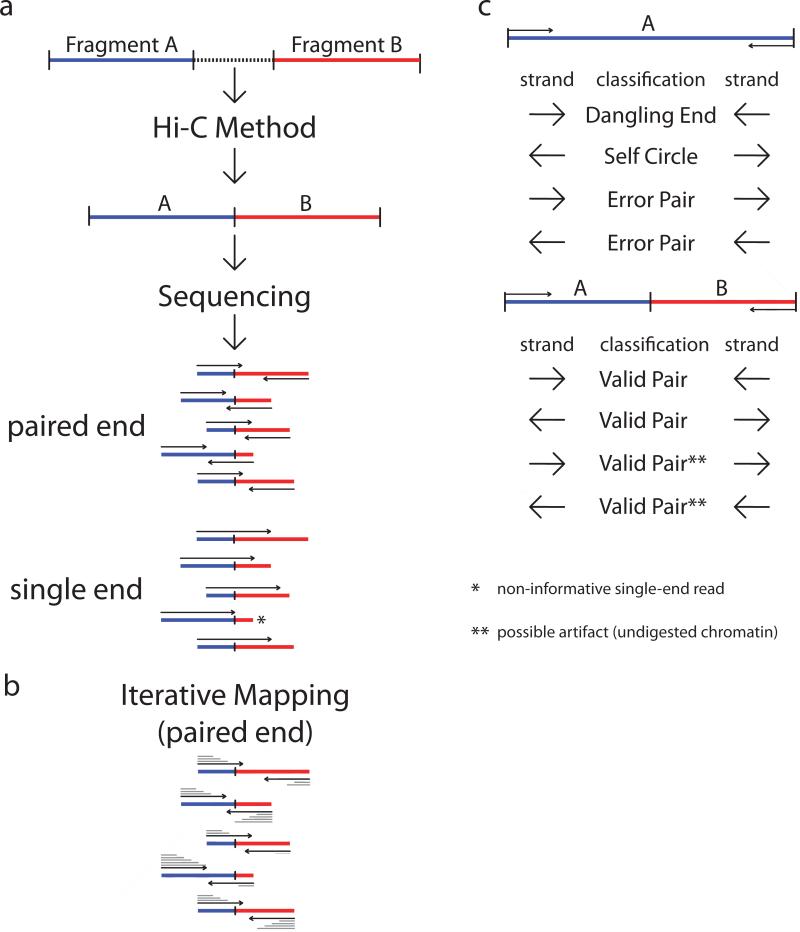
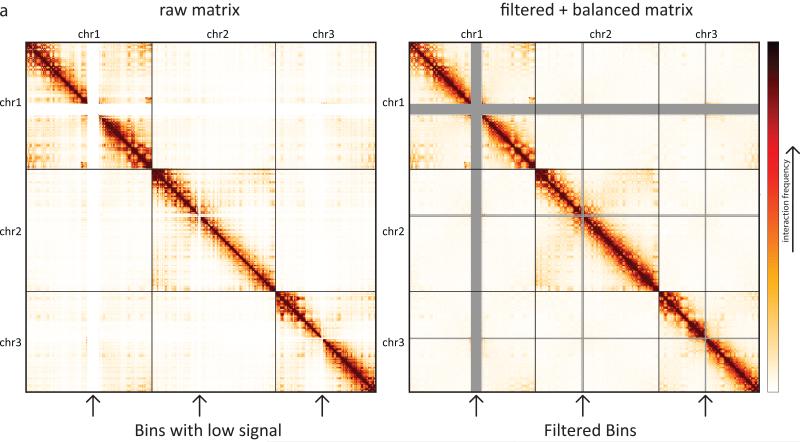
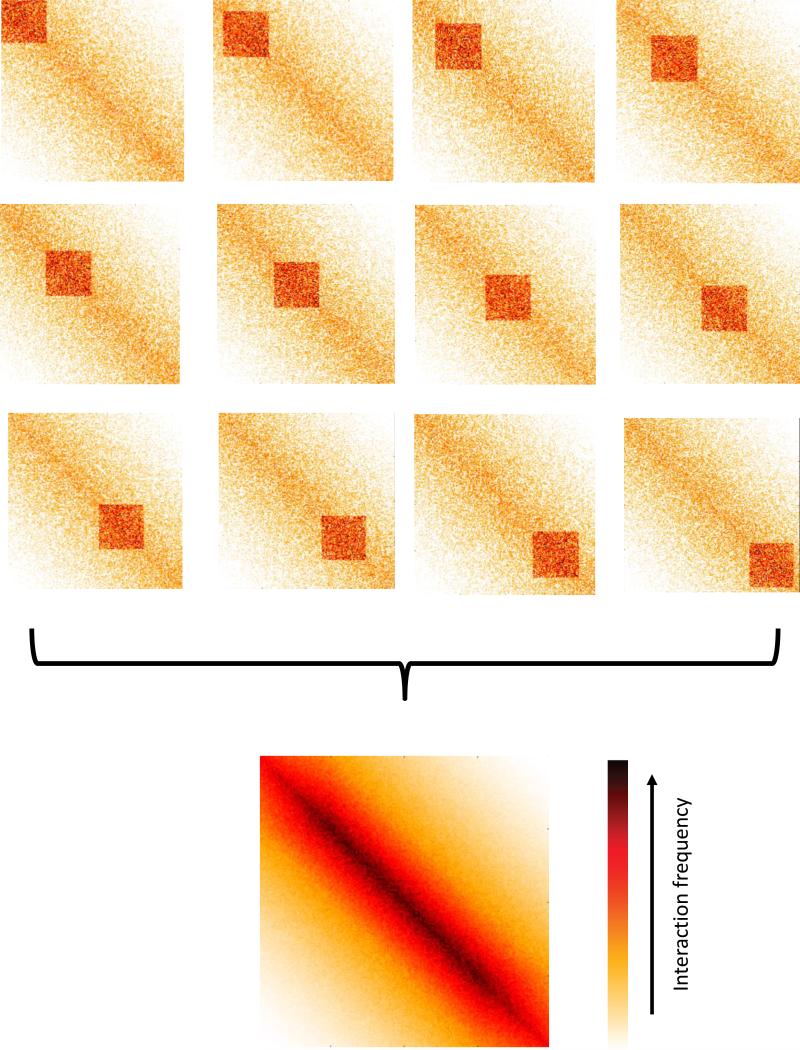
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
