

Pancreatic cell plasticity and cancer initiation induced by oncogenic Kras is completely dependent on wild-type PI 3-kinase p110α
- PMID: 25452273
- PMCID: PMC4248293
- DOI: 10.1101/gad.249409.114
Pancreatic cell plasticity and cancer initiation induced by oncogenic Kras is completely dependent on wild-type PI 3-kinase p110α
Abstract
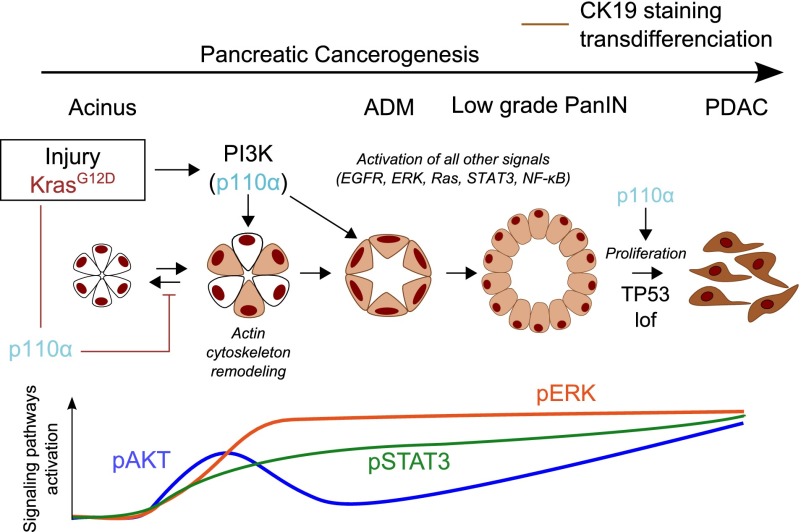
Increased PI 3-kinase (PI3K) signaling in pancreatic ductal adenocarcinoma (PDAC) correlates with poor prognosis, but the role of class I PI3K isoforms during its induction remains unclear. Using genetically engineered mice and pharmacological isoform-selective inhibitors, we found that the p110α PI3K isoform is a major signaling enzyme for PDAC development induced by a combination of genetic and nongenetic factors. Inactivation of this single isoform blocked the irreversible transition of exocrine acinar cells into pancreatic preneoplastic ductal lesions by oncogenic Kras and/or pancreatic injury. Hitting the other ubiquitous isoform, p110β, did not prevent preneoplastic lesion initiation. p110α signaling through small GTPase Rho and actin cytoskeleton controls the reprogramming of acinar cells and regulates cell morphology in vivo and in vitro. Finally, p110α was necessary for pancreatic ductal cancers to arise from Kras-induced preneoplastic lesions by increasing epithelial cell proliferation in the context of mutated p53. Here we identify an in vivo context in which p110α cellular output differs depending on the epithelial transformation stage and demonstrate that the PI3K p110α is required for PDAC induced by oncogenic Kras, the key driver mutation of PDAC. These data are critical for a better understanding of the development of this lethal disease that is currently without efficient treatment.
Keywords: PI3K isoforms; genetic mouse models; oncogenes; pancreas; signaling; transdifferentiation.
© 2014 Baer et al.; Published by Cold Spring Harbor Laboratory Press.
Figures
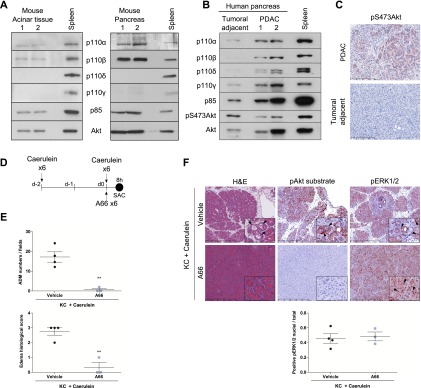
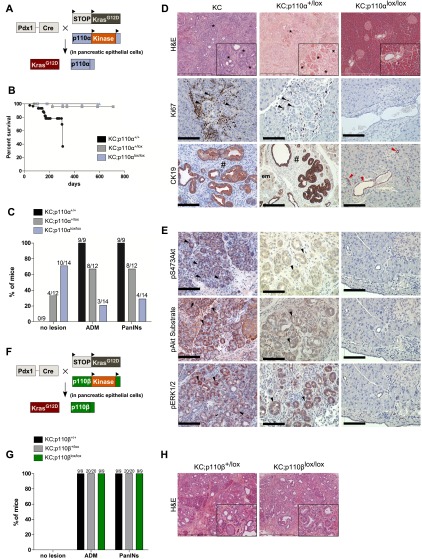
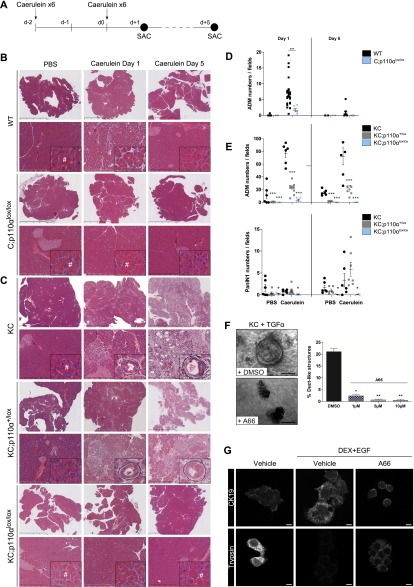
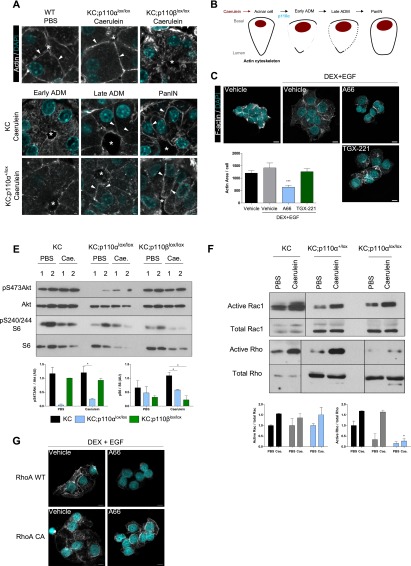
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous