

Conformational dynamics of a crystalline protein from microsecond-scale molecular dynamics simulations and diffuse X-ray scattering
- PMID: 25453071
- PMCID: PMC4273327
- DOI: 10.1073/pnas.1416744111
Conformational dynamics of a crystalline protein from microsecond-scale molecular dynamics simulations and diffuse X-ray scattering
Abstract
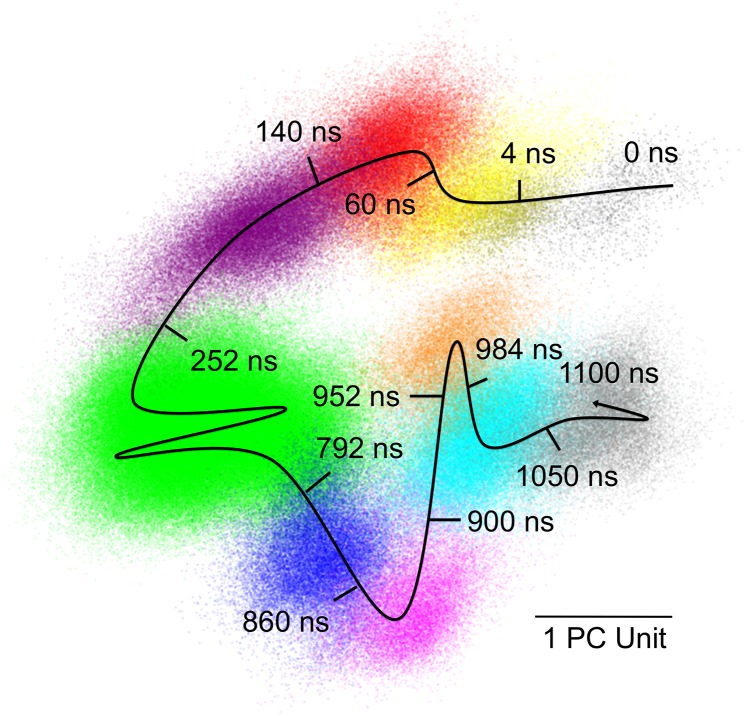
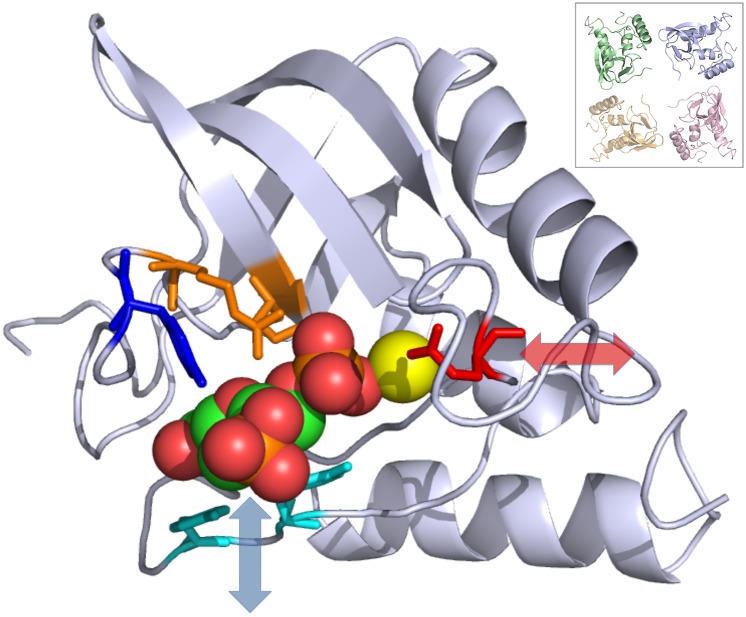
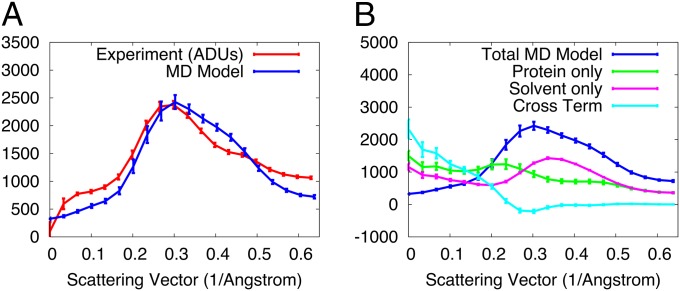
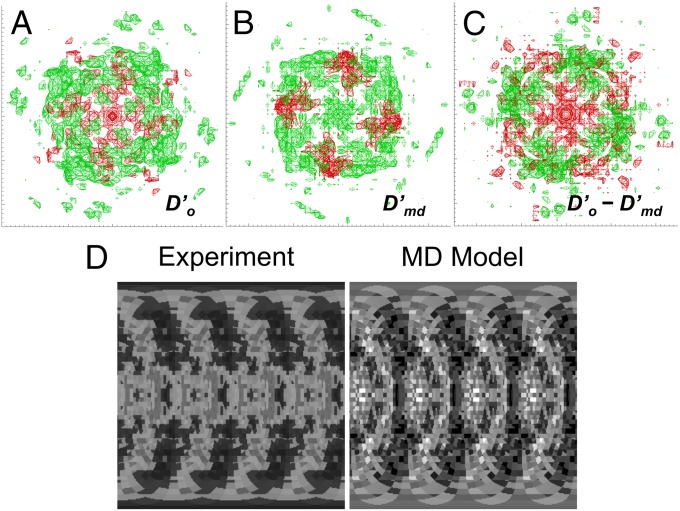
X-ray diffraction from protein crystals includes both sharply peaked Bragg reflections and diffuse intensity between the peaks. The information in Bragg scattering is limited to what is available in the mean electron density. The diffuse scattering arises from correlations in the electron density variations and therefore contains information about collective motions in proteins. Previous studies using molecular-dynamics (MD) simulations to model diffuse scattering have been hindered by insufficient sampling of the conformational ensemble. To overcome this issue, we have performed a 1.1-μs MD simulation of crystalline staphylococcal nuclease, providing 100-fold more sampling than previous studies. This simulation enables reproducible calculations of the diffuse intensity and predicts functionally important motions, including transitions among at least eight metastable states with different active-site geometries. The total diffuse intensity calculated using the MD model is highly correlated with the experimental data. In particular, there is excellent agreement for the isotropic component of the diffuse intensity, and substantial but weaker agreement for the anisotropic component. Decomposition of the MD model into protein and solvent components indicates that protein-solvent interactions contribute substantially to the overall diffuse intensity. We conclude that diffuse scattering can be used to validate predictions from MD simulations and can provide information to improve MD models of protein motions.
Keywords: diffuse scattering; molecular-dynamics simulation; protein crystallography; protein dynamics; staphylococcal nuclease.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Austin RH, et al. Dynamics of carbon monoxide binding by heme proteins. Science. 1973;181(4099):541–543. - PubMed
-
- Weber G. Ligand binding and internal equilibria in proteins. Biochemistry. 1972;11(5):864–878. - PubMed
-
- Frauenfelder H, Parak F, Young RD. Conformational substates in proteins. Annu Rev Biophys Biophys Chem. 1988;17:451–479. - PubMed
-
- Frauenfelder H, Petsko GA, Tsernoglou D. Temperature-dependent X-ray diffraction as a probe of protein structural dynamics. Nature. 1979;280(5723):558–563. - PubMed
-
- Doscher MS, Richards FM. The activity of an enzyme in the crystalline state: Ribonuclease S. J Biol Chem. 1963;238(7):2393–2398.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
