

Recessive cardiac phenotypes in induced pluripotent stem cell models of Jervell and Lange-Nielsen syndrome: disease mechanisms and pharmacological rescue
- PMID: 25453094
- PMCID: PMC4273331
- DOI: 10.1073/pnas.1419553111
Recessive cardiac phenotypes in induced pluripotent stem cell models of Jervell and Lange-Nielsen syndrome: disease mechanisms and pharmacological rescue
Abstract
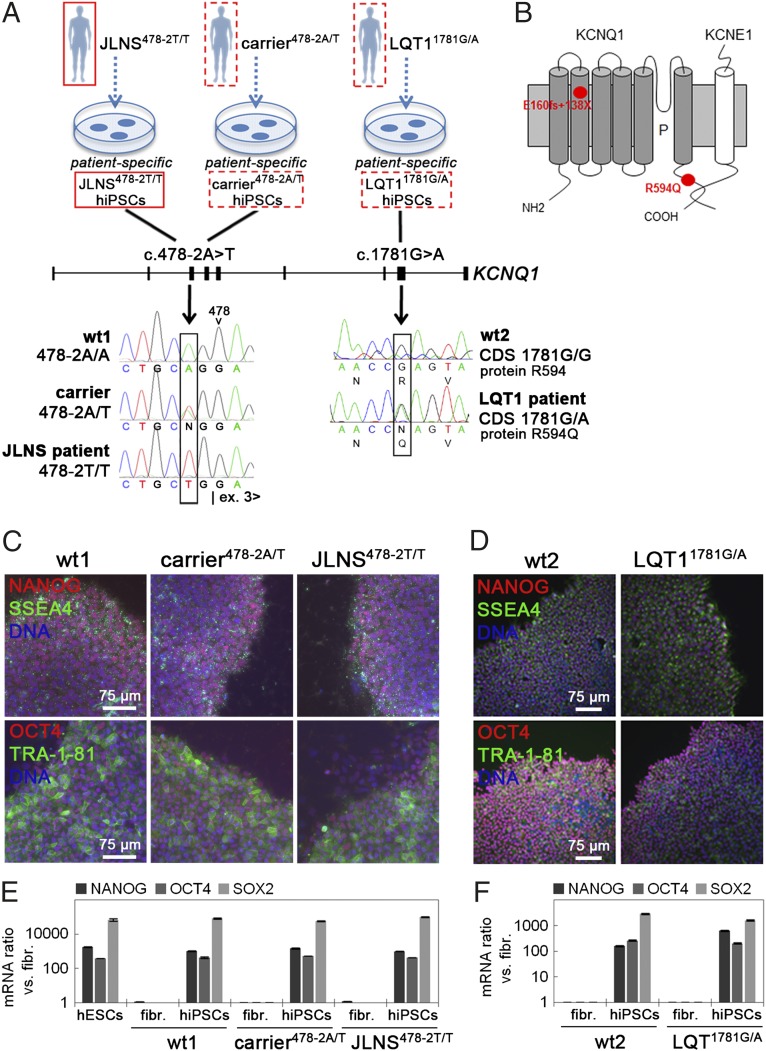
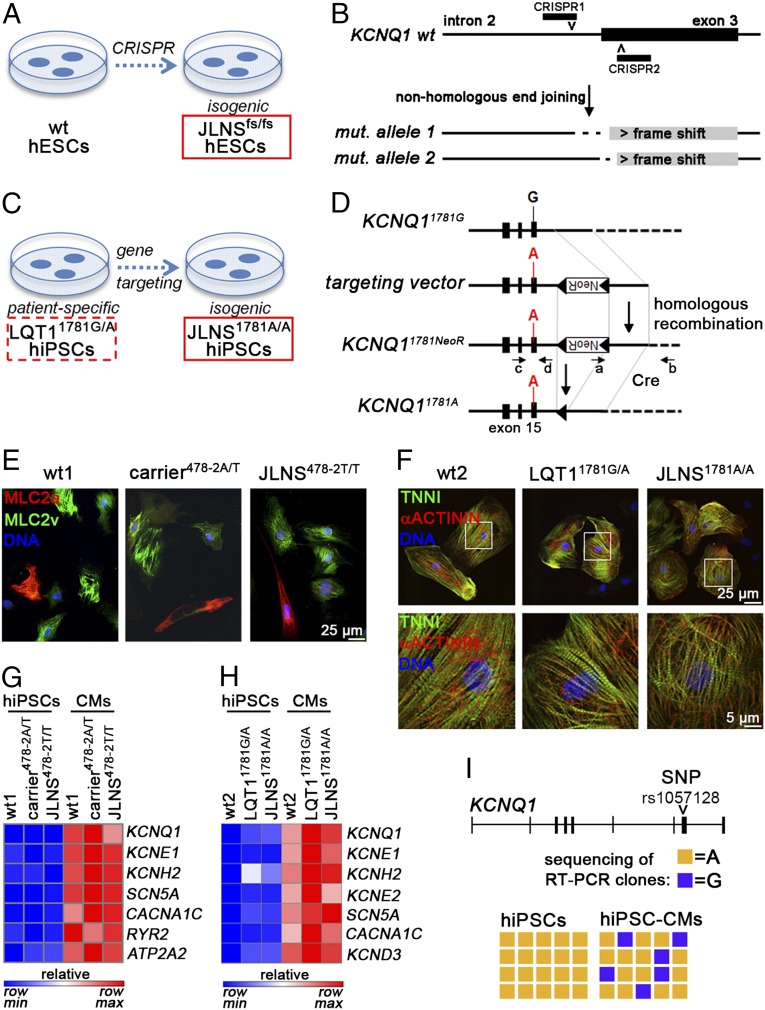
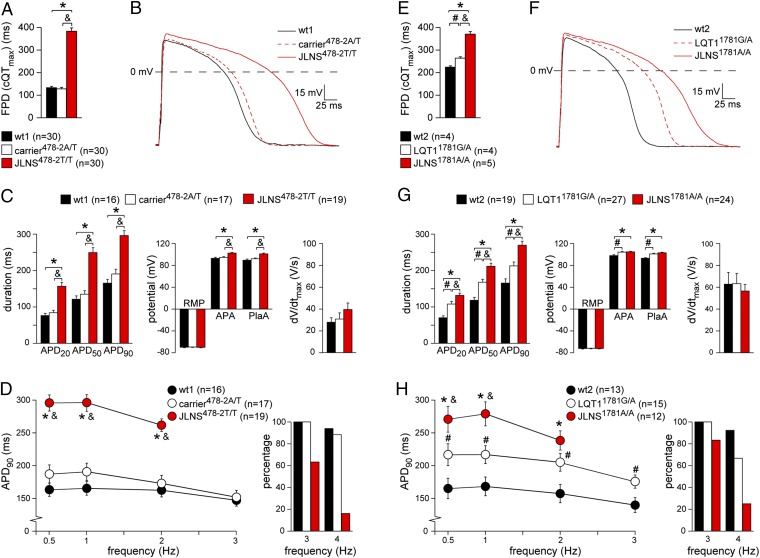
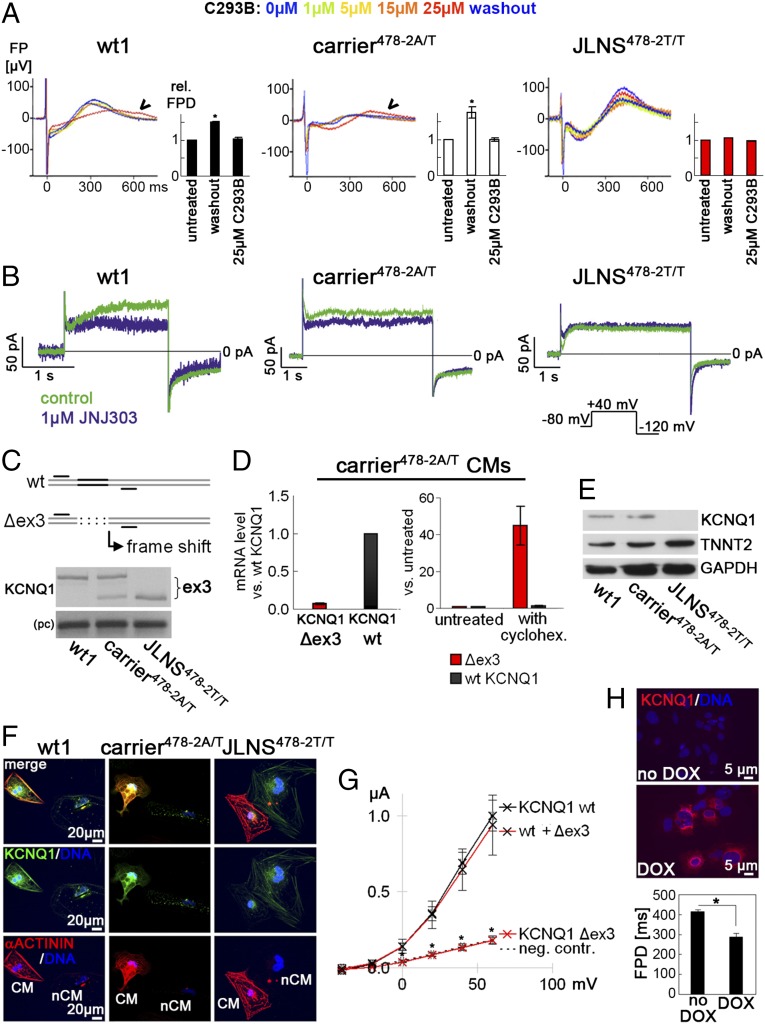
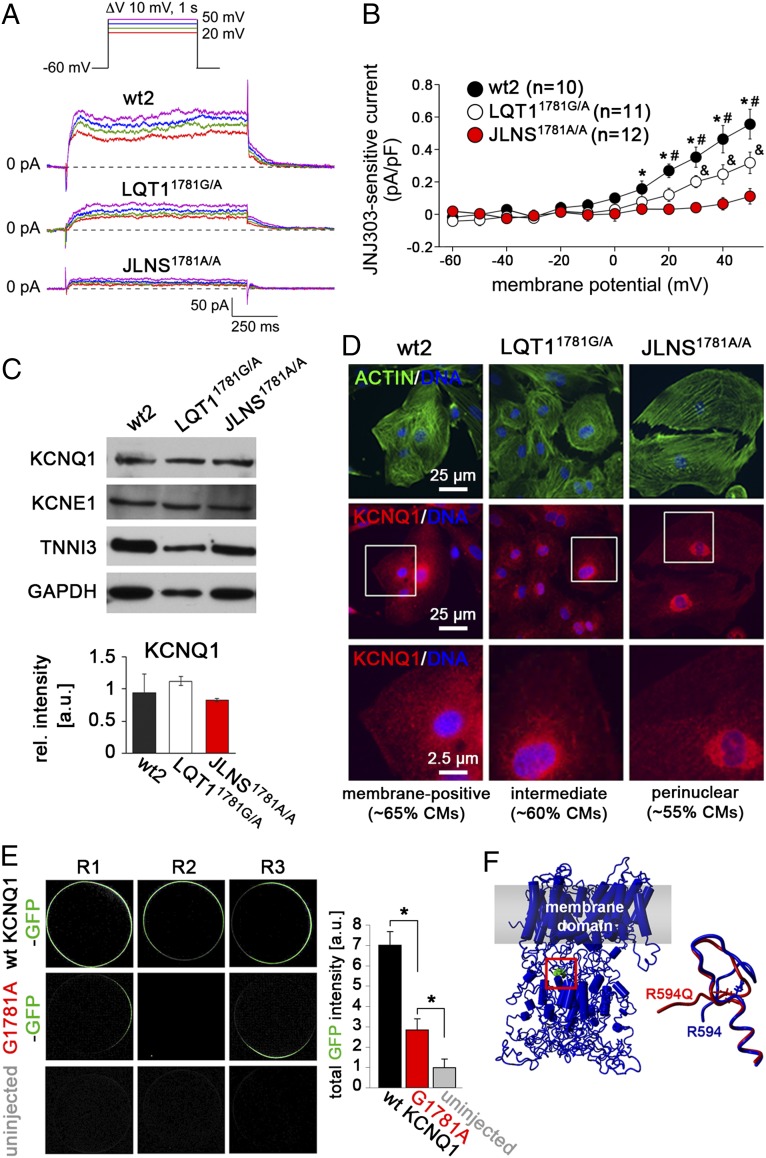
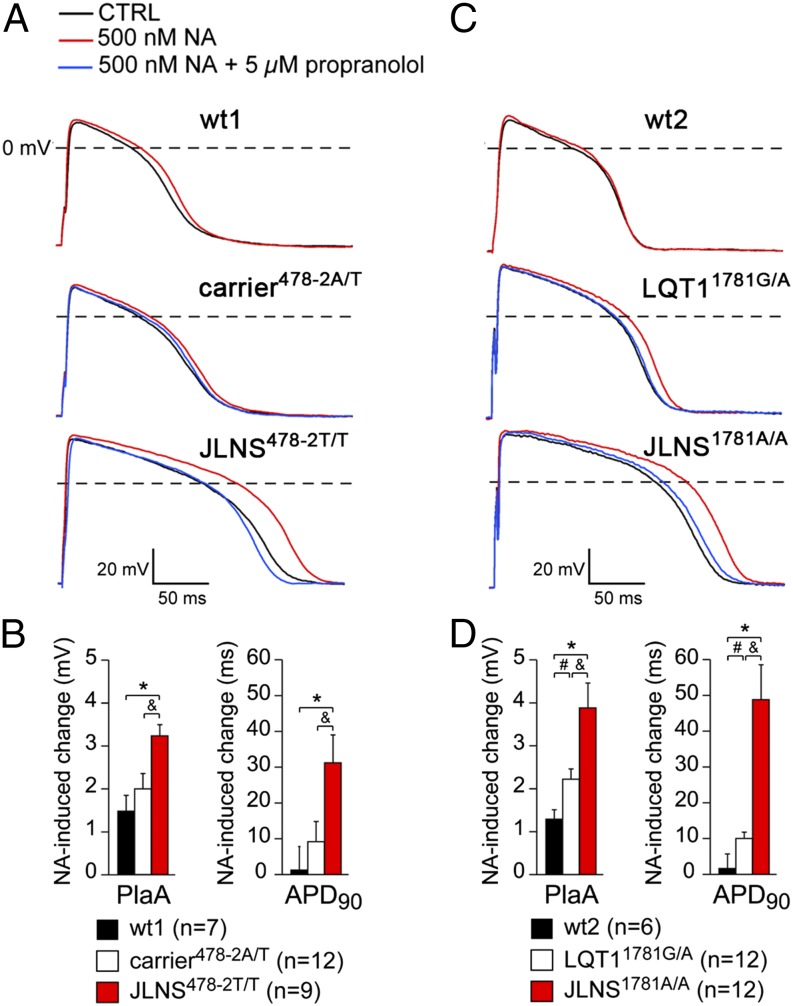
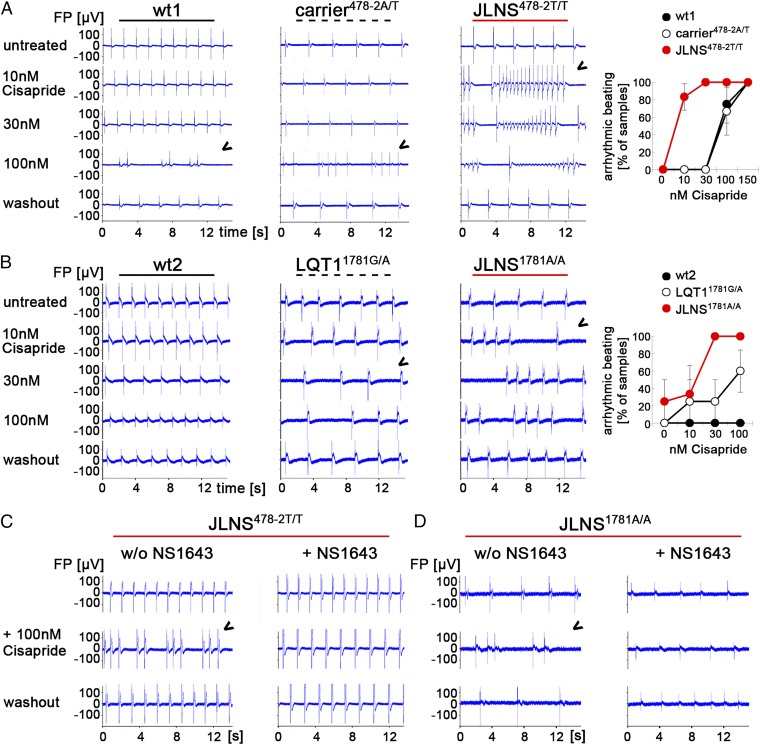
Jervell and Lange-Nielsen syndrome (JLNS) is one of the most severe life-threatening cardiac arrhythmias. Patients display delayed cardiac repolarization, associated high risk of sudden death due to ventricular tachycardia, and congenital bilateral deafness. In contrast to the autosomal dominant forms of long QT syndrome, JLNS is a recessive trait, resulting from homozygous (or compound heterozygous) mutations in KCNQ1 or KCNE1. These genes encode the α and β subunits, respectively, of the ion channel conducting the slow component of the delayed rectifier K(+) current, IKs. We used complementary approaches, reprogramming patient cells and genetic engineering, to generate human induced pluripotent stem cell (hiPSC) models of JLNS, covering splice site (c.478-2A>T) and missense (c.1781G>A) mutations, the two major classes of JLNS-causing defects in KCNQ1. Electrophysiological comparison of hiPSC-derived cardiomyocytes (CMs) from homozygous JLNS, heterozygous, and wild-type lines recapitulated the typical and severe features of JLNS, including pronounced action and field potential prolongation and severe reduction or absence of IKs. We show that this phenotype had distinct underlying molecular mechanisms in the two sets of cell lines: the previously unidentified c.478-2A>T mutation was amorphic and gave rise to a strictly recessive phenotype in JLNS-CMs, whereas the missense c.1781G>A lesion caused a gene dosage-dependent channel reduction at the cell membrane. Moreover, adrenergic stimulation caused action potential prolongation specifically in JLNS-CMs. Furthermore, sensitivity to proarrhythmic drugs was strongly enhanced in JLNS-CMs but could be pharmacologically corrected. Our data provide mechanistic insight into distinct classes of JLNS-causing mutations and demonstrate the potential of hiPSC-CMs in drug evaluation.
Keywords: Jervell and Lange-Nielsen syndrome; KCNQ1; disease modeling; human induced pluripotent stem cells; long QT syndrome.
Conflict of interest statement
Conflict of interest statement: C.L.M. is cofounder and advisor of Pluriomics.
Figures
Comment in
-
Reply to Christ et al.: LQT1 and JLNS phenotypes in hiPSC-derived cardiomyocytes are due to KCNQ1 mutations.Proc Natl Acad Sci U S A. 2015 Apr 21;112(16):E1969. doi: 10.1073/pnas.1503762112. Epub 2015 Mar 20. Proc Natl Acad Sci U S A. 2015. PMID: 25795241 Free PMC article. No abstract available.
-
LQT1-phenotypes in hiPSC: Are we measuring the right thing?Proc Natl Acad Sci U S A. 2015 Apr 21;112(16):E1968. doi: 10.1073/pnas.1503347112. Epub 2015 Mar 20. Proc Natl Acad Sci U S A. 2015. PMID: 25795242 Free PMC article. No abstract available.
References
-
- Jervell A, Lange-Nielsen F. Congenital deaf-mutism, functional heart disease with prolongation of the Q-T interval and sudden death. Am Heart J. 1957;54(1):59–68. - PubMed
-
- Schwartz PJ, et al. The Jervell and Lange-Nielsen syndrome: Natural history, molecular basis, and clinical outcome. Circulation. 2006;113(6):783–790. - PubMed
-
- Jespersen T, Grunnet M, Olesen SP. The KCNQ1 potassium channel: From gene to physiological function. Physiology (Bethesda) 2005;20:408–416. - PubMed
-
- Schulze-Bahr E, et al. KCNE1 mutations cause jervell and Lange-Nielsen syndrome. Nat Genet. 1997;17(3):267–268. - PubMed
-
- Romano C, Gemme G, Pongiglione R. [Rare cardiac arrythmias of the pediatric age. Ii. syncopal attacks due to paroxysmal ventricular fibrillation. (Presentation of 1st case in Italian pediatric literature)] Clin Pediatr (Bologna) 1963;45:656–683. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
