

Increased extracellular pressure stimulates tumor proliferation by a mechanosensitive calcium channel and PKC-β
- PMID: 25454347
- PMCID: PMC4487881
- DOI: 10.1016/j.molonc.2014.10.008
Increased extracellular pressure stimulates tumor proliferation by a mechanosensitive calcium channel and PKC-β
Abstract
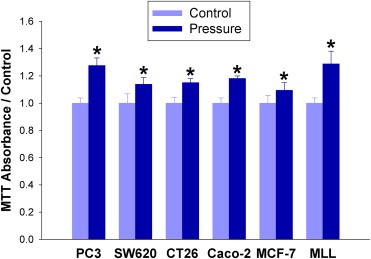
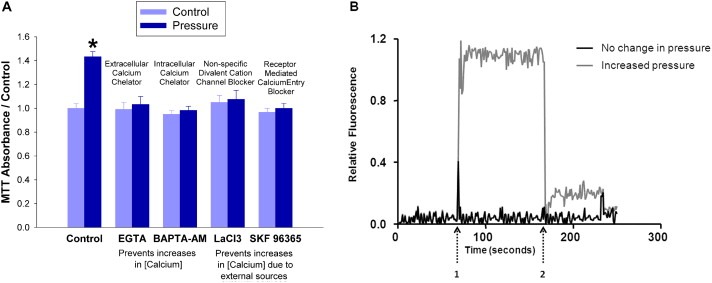
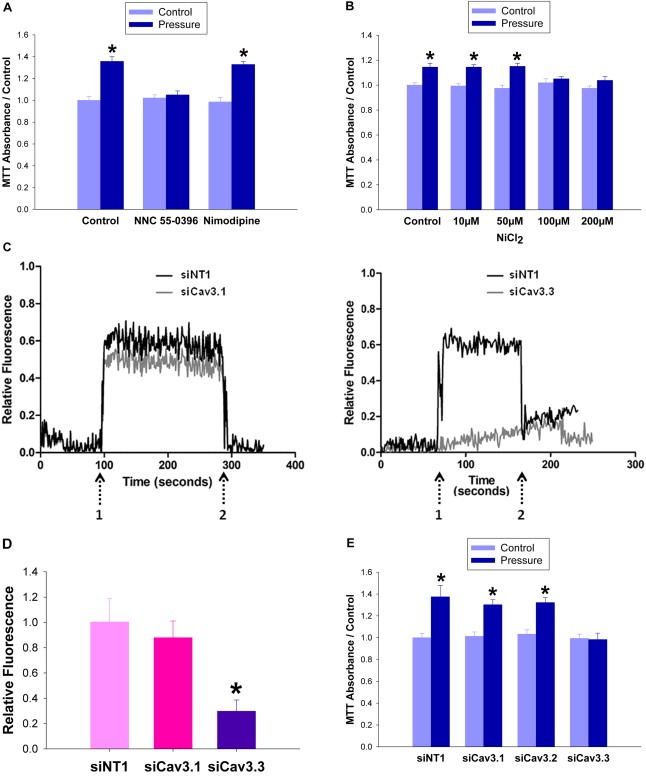
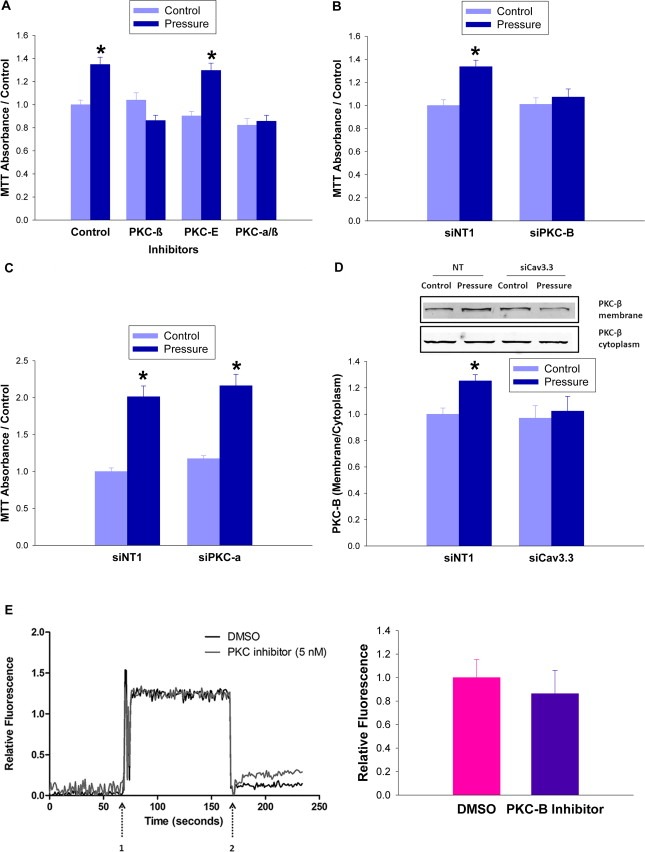
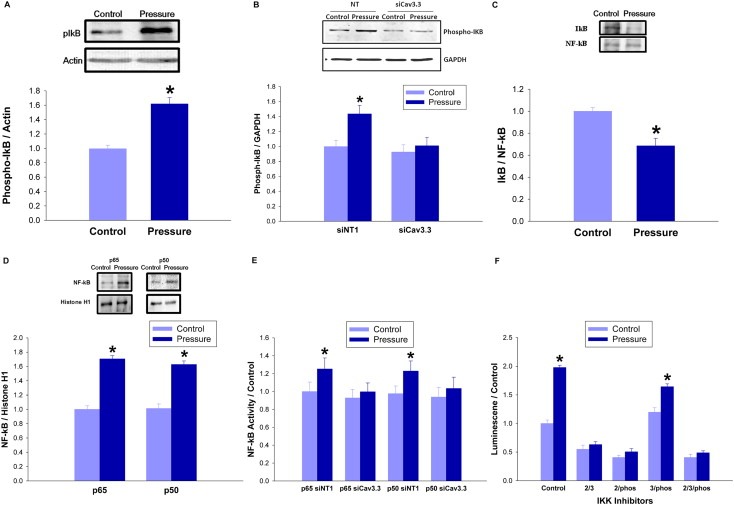
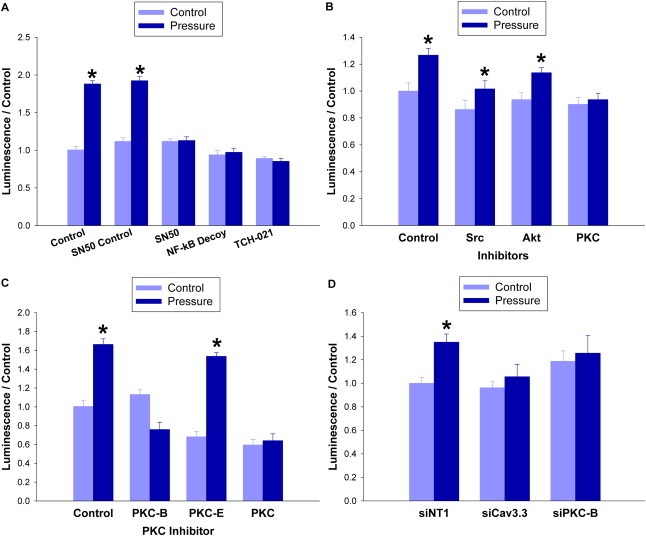
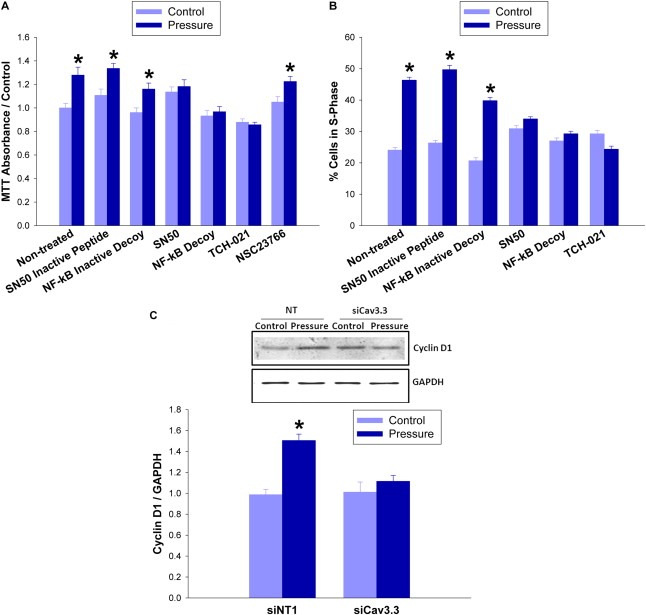
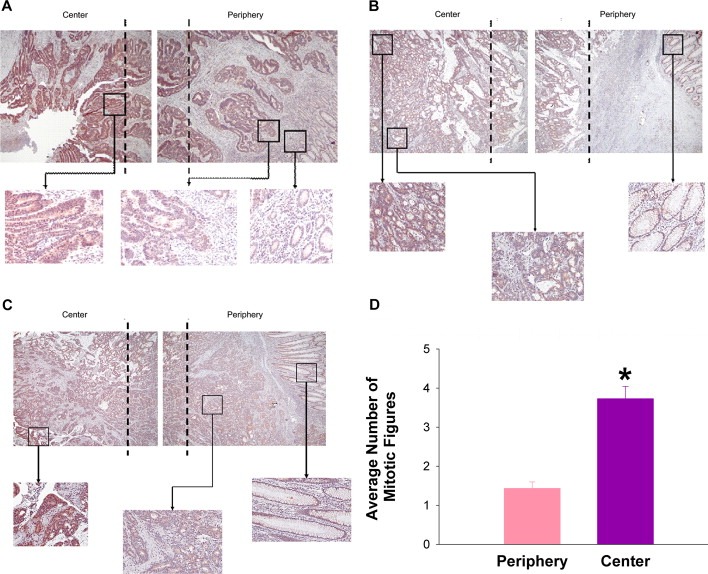
Large tumors exhibit high interstitial pressure heightened by growth against the constraining stroma. Such pressures could stimulate tumor proliferation via a mechanosensitive ion channel. We studied the effects of 0-80 mmHg increased extracellular pressure for 24 h on proliferation of SW620, Caco-2, and CT-26 colon; MCF-7 breast; and MLL and PC3 prostate cancer cells, and delineated its mechanism in SW620 cells with specific inhibitors and siRNA. Finally, we compared NF-kB, phospho-IkB and cyclin D1 immunoreactivity in the high pressure centers and low pressure peripheries of human tumors. Pressure-stimulated proliferation in all cells. Pressure-driven SW620 proliferation required calcium influx via the T-type Ca(2+) channel Cav3.3, which stimulated PKC-β to invoke the IKK-IkB-NF-kB pathway to increase proliferation and S-phase fraction. The mitotic index and immunoreactivity of NF-kB, phospho-IkB, and cyclin D1 in the center of 28 large human colon, lung, and head and neck tumors exceeded that in tumor peripheries. Extracellular pressure increases [Ca(2+)]i via Cav3.3, driving a PKC-β- IKK- IkB-NF-kB pathway that stimulates cancer cell proliferation. Rapid proliferation in large stiff tumors may increase intratumoral pressure, activating this pathway to stimulate further proliferation in a feedback cycle that potentiates tumor growth. Targeting this pathway may inhibit proliferation in large unresectable tumors.
Keywords: Calcium channels; Cav3.3; NF-kB; PKC; Pressure; Proliferation.
Copyright © 2014 Federation of European Biochemical Societies. Published by Elsevier B.V. All rights reserved.
Figures
References
-
- Ariffin, A.B. , Forde, P.F. , Jahangeer, S. , Soden, D.M. , Hinchion, J. , 2014. Releasing pressure in tumors: what do we know so far and where do we go from here? A review. Cancer Res. 74, 2655–2662. - PubMed
-
- Basson, M.D. , 2008. An intracellular signal pathway that regulates cancer cell adhesion in response to extracellular forces. Cancer Res. 68, 2–4. - PubMed
-
- Basson, M.D. , Hong, F. , 1995. Modulation of human CACO-2 intestinal epithelial cell phenotype by protein kinase C inhibitors. Cell Biol. Int. 19, 1025–1032. - PubMed
-
- Bertolesi, G.E. , Shi, C. , Elbaum, L. , Jollimore, C. , Rozenberg, G. , Barnes, S. , Kelly, M.E. , 2002. The Ca(2+) channel antagonists mibefradil and pimozide inhibit cell growth via different cytotoxic mechanisms. Mol. Pharmacol. 62, 210–219. - PubMed
-
- Bevan, R.D. , 1976. An autoradiographic and pathological study of cellular proliferation in rabbit arteries correlated with an increase in arterial pressure. Blood Vessels. 13, 100–128. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
