

Regulation of Stat5 by FAK and PAK1 in Oncogenic FLT3- and KIT-Driven Leukemogenesis
- PMID: 25456130
- PMCID: PMC4380442
- DOI: 10.1016/j.celrep.2014.10.039
Regulation of Stat5 by FAK and PAK1 in Oncogenic FLT3- and KIT-Driven Leukemogenesis
Abstract
Oncogenic mutations of FLT3 and KIT receptors are associated with poor survival in patients with acute myeloid leukemia (AML) and myeloproliferative neoplasms (MPNs), and currently available drugs are largely ineffective. Although Stat5 has been implicated in regulating several myeloid and lymphoid malignancies, how precisely Stat5 regulates leukemogenesis, including its nuclear translocation to induce gene transcription, is poorly understood. In leukemic cells, we show constitutive activation of focal adhesion kinase (FAK) whose inhibition represses leukemogenesis. Downstream of FAK, activation of Rac1 is regulated by RacGEF Tiam1, whose inhibition prolongs the survival of leukemic mice. Inhibition of the Rac1 effector PAK1 prolongs the survival of leukemic mice in part by inhibiting the nuclear translocation of Stat5. These results reveal a leukemic pathway involving FAK/Tiam1/Rac1/PAK1 and demonstrate an essential role for these signaling molecules in regulating the nuclear translocation of Stat5 in leukemogenesis.
Copyright © 2014 The Authors. Published by Elsevier Inc. All rights reserved.
Figures

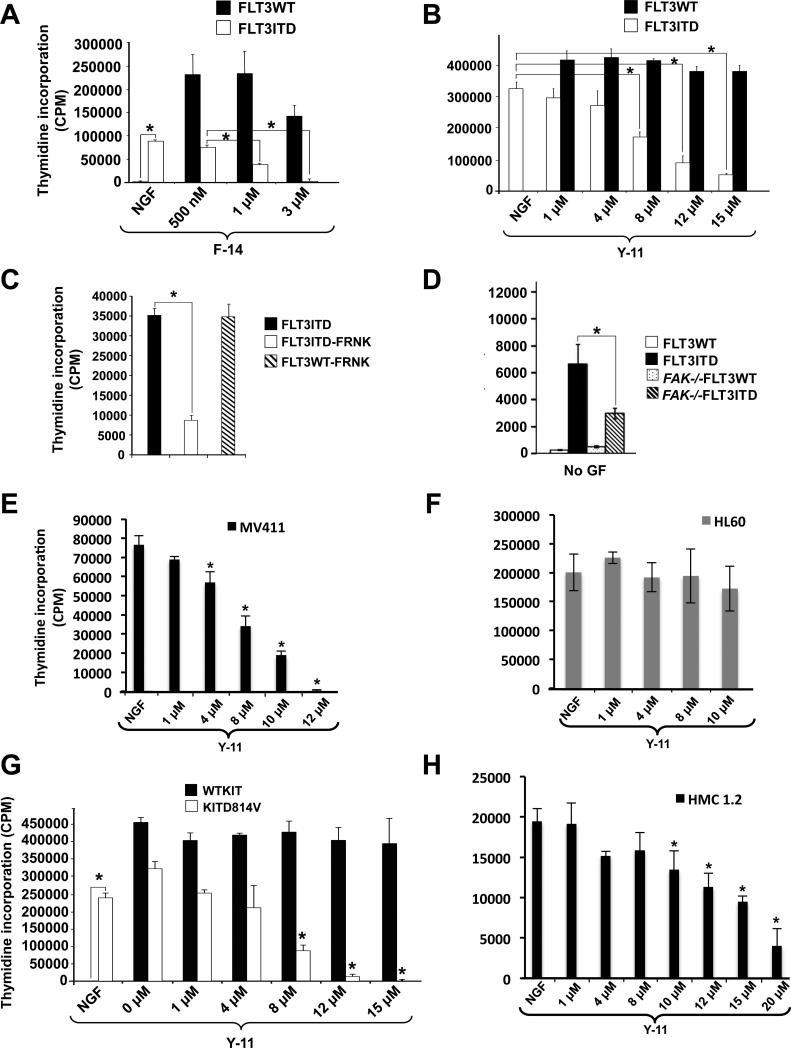




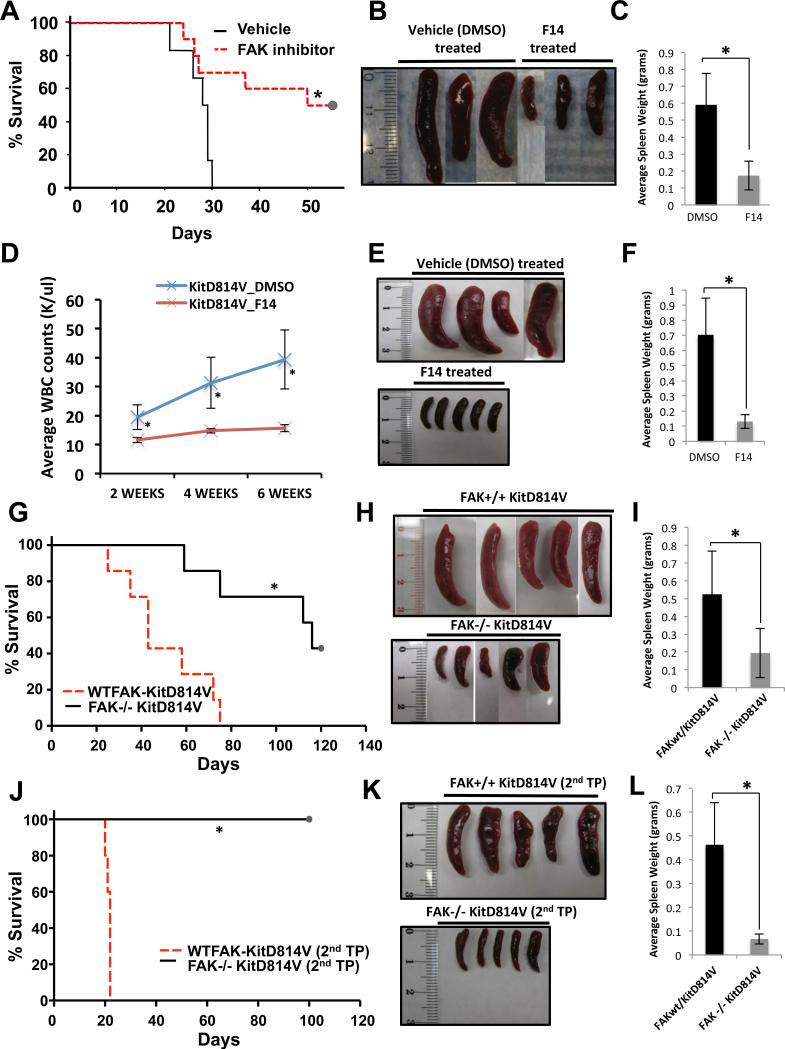
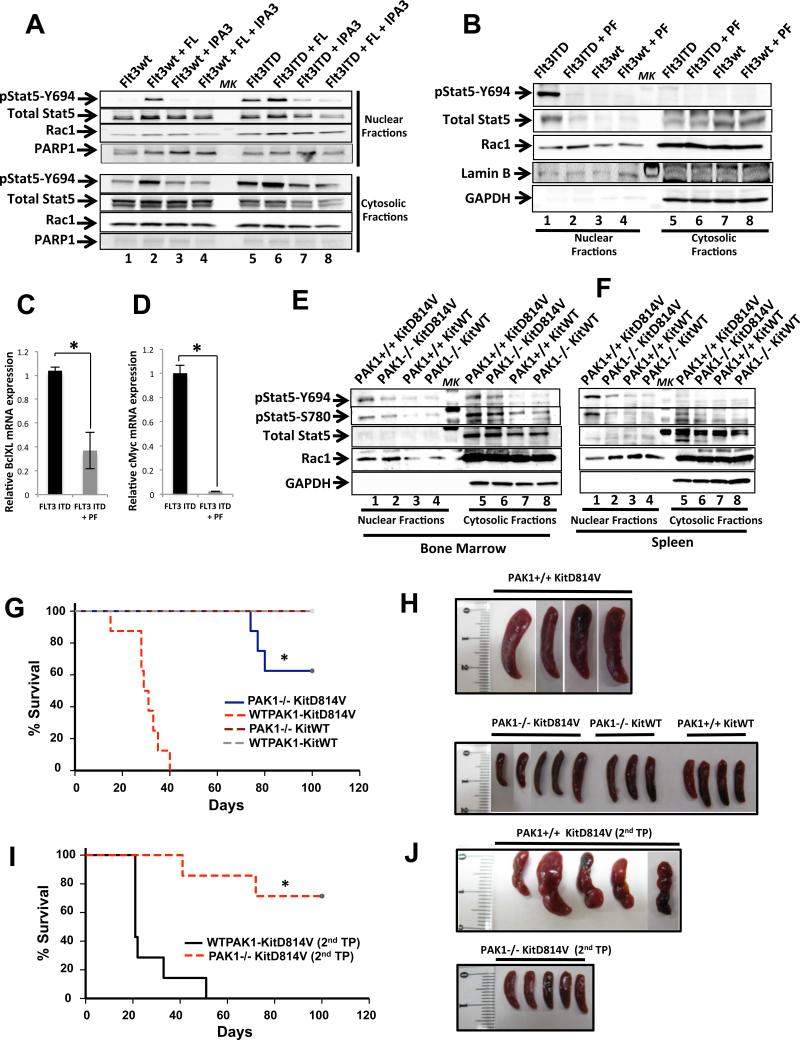
References
-
- Baumgartner C, Cerny-Reiterer S, Sonneck K, Mayerhofer M, Gleixner KV, Fritz R, Kerenyi M, Boudot C, Gouilleux F, Kornfeld JW, et al. Expression of activated STAT5 in neoplastic mast cells in systemic mastocytosis: subcellular distribution and role of the transforming oncoprotein KIT D816V. The American journal of pathology. 2009;175:2416–2429. - PMC - PubMed
-
- Beghini A, Ripamonti CB, Cairoli R, Cazzaniga G, Colapietro P, Elice F, Nadali G, Grillo G, Haas OA, Biondi A, et al. KIT activating mutations: incidence in adult and pediatric acute myeloid leukemia, and identification of an internal tandem duplication. Haematologica. 2004;89:920–925. - PubMed
-
- Benekli M, Baer MR, Baumann H, Wetzler M. Signal transducer and activator of transcription proteins in leukemias. Blood. 2003;101:2940–2954. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
