

Whole genome de novo assemblies of three divergent strains of rice, Oryza sativa, document novel gene space of aus and indica
- PMID: 25468217
- PMCID: PMC4268812
- DOI: 10.1186/PREACCEPT-2784872521277375
Whole genome de novo assemblies of three divergent strains of rice, Oryza sativa, document novel gene space of aus and indica
Abstract
Background: The use of high throughput genome-sequencing technologies has uncovered a large extent of structural variation in eukaryotic genomes that makes important contributions to genomic diversity and phenotypic variation. When the genomes of different strains of a given organism are compared, whole genome resequencing data are typically aligned to an established reference sequence. However, when the reference differs in significant structural ways from the individuals under study, the analysis is often incomplete or inaccurate.
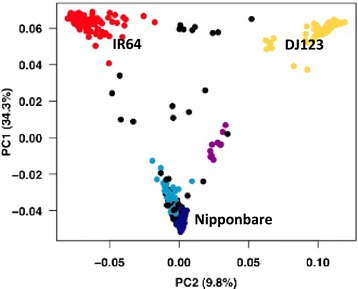
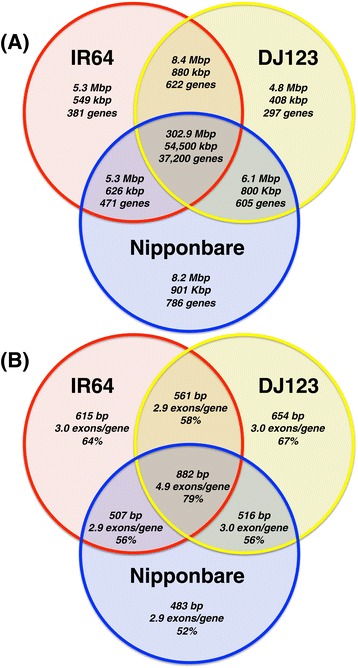
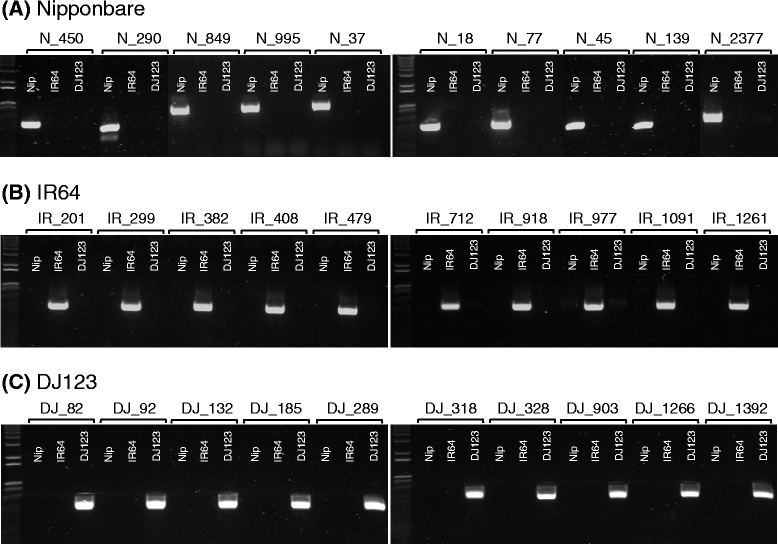
Results: Here, we use rice as a model to demonstrate how improvements in sequencing and assembly technology allow rapid and inexpensive de novo assembly of next generation sequence data into high-quality assemblies that can be directly compared using whole genome alignment to provide an unbiased assessment. Using this approach, we are able to accurately assess the "pan-genome" of three divergent rice varieties and document several megabases of each genome absent in the other two.
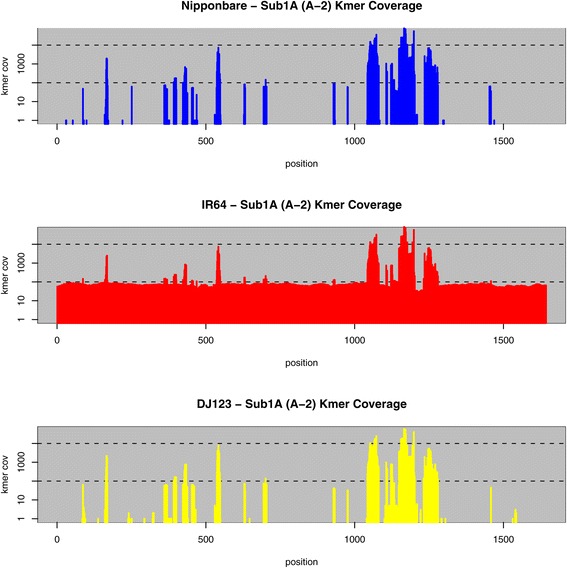
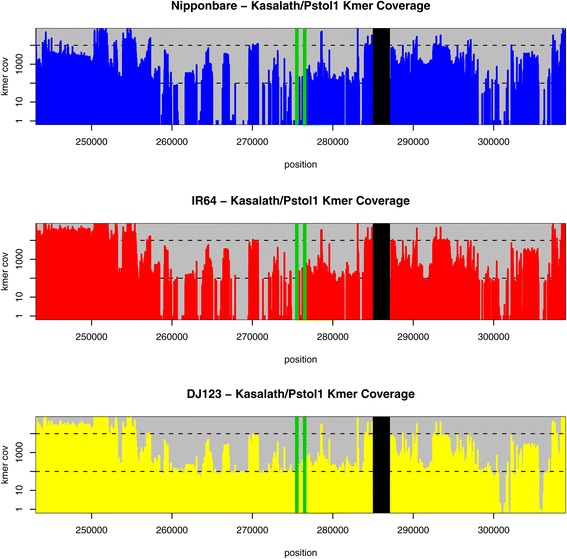
Conclusions: Many of the genome-specific loci are annotated to contain genes, reflecting the potential for new biological properties that would be missed by standard reference-mapping approaches. We further provide a detailed analysis of several loci associated with agriculturally important traits, including the S5 hybrid sterility locus, the Sub1 submergence tolerance locus, the LRK gene cluster associated with improved yield, and the Pup1 cluster associated with phosphorus deficiency, illustrating the utility of our approach for biological discovery. All of the data and software are openly available to support further breeding and functional studies of rice and other species.
Figures
References
-
- Huang X, Kurata N, Wei X, Wang ZX, Wang A, Zhao Q, Zhao Y, Liu K, Lu H, Li W, Guo Y, Lu Y, Zhou C, Fan D, Weng Q, Zhu C, Huang T, Zhang L, Wang Y, Feng L, Furuumi H, Kubo T, Miyabayashi T, Yuan X, Xu Q, Dong G, Zhan Q, Li C, Fujiyama A, Toyoda A, et al. A map of rice genome variation reveals the origin of cultivated rice. Nature. 2012;490:497–501. doi: 10.1038/nature11532. - DOI - PMC - PubMed
-
- Zhao KY, Wright M, Kimball J, Eizenga G, McClung A, Kovach M, Tyagi W, Ali ML, Tung CW, Reynolds A, Bustamante CD, McCouch SR. Genomic diversity and introgression in O. sativa reveal the impact of domestication and breeding on the rice genome. Plos One. 2010;5:e10780. doi: 10.1371/journal.pone.0010780. - DOI - PMC - PubMed
-
- Boyko AR, Quignon P, Li L, Schoenebeck JJ, Degenhardt JD, Lohmueller KE, Zhao K, Brisbin A, Parker HG, vonHoldt BM, Cargill M, Auton A, Reynolds A, Elkahloun AG, Castelhano M, Mosher DS, Sutter NB, Johnson GS, Novembre J, Hubisz MJ, Siepel A, Wayne RK, Bustamante CD, Ostrander EA. A simple genetic architecture underlies morphological variation in dogs. PLoS Biol. 2010;8:e1000451. doi: 10.1371/journal.pbio.1000451. - DOI - PMC - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
