

Assessing copy number alterations in targeted, amplicon-based next-generation sequencing data
- PMID: 25468433
- PMCID: PMC5707205
- DOI: 10.1016/j.jmoldx.2014.09.008
Assessing copy number alterations in targeted, amplicon-based next-generation sequencing data
Abstract
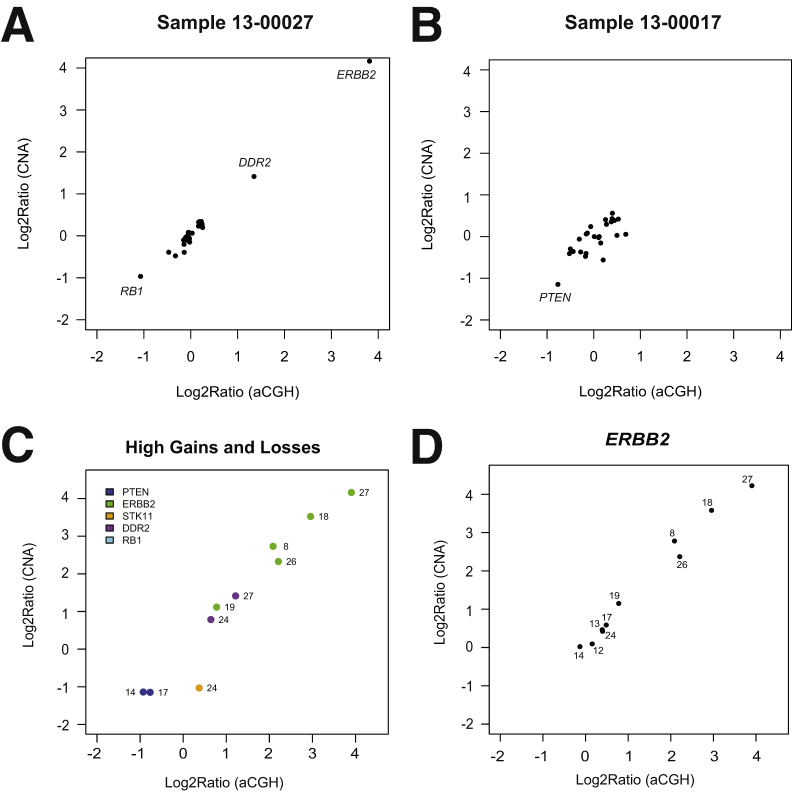
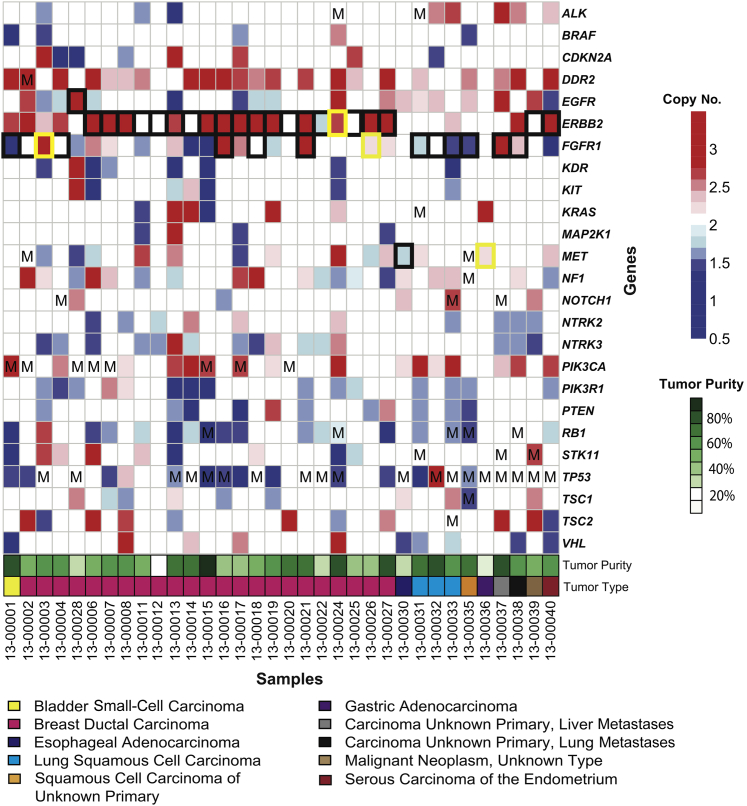
Changes in gene copy number are important in the setting of precision medicine. Recent studies have established that copy number alterations (CNAs) can be detected in sequencing libraries prepared by hybridization-capture, but there has been comparatively little attention given to CNA assessment in amplicon-based libraries prepared by PCR. In this study, we developed an algorithm for detecting CNAs in amplicon-based sequencing data. CNAs determined from the algorithm mirrored those from a hybridization-capture library. In addition, analysis of 14 pairs of matched normal and breast carcinoma tissues revealed that sequence data pooled from normal samples could be substituted for a matched normal tissue without affecting the detection of clinically relevant CNAs (>|2| copies). Comparison of CNAs identified by array comparative genomic hybridization and amplicon-based libraries across 10 breast carcinoma samples showed an excellent correlation. The CNA algorithm also compared favorably with fluorescence in situ hybridization, with agreement in 33 of 38 assessments across four different genes. Factors that influenced the detection of CNAs included the number of amplicons per gene, the average read depth, and, most important, the proportion of tumor within the sample. Our results show that CNAs can be identified in amplicon-based targeted sequencing data, and that their detection can be optimized by ensuring adequate tumor content and read coverage.
Copyright © 2015 American Society for Investigative Pathology and the Association for Molecular Pathology. Published by Elsevier Inc. All rights reserved.
Figures
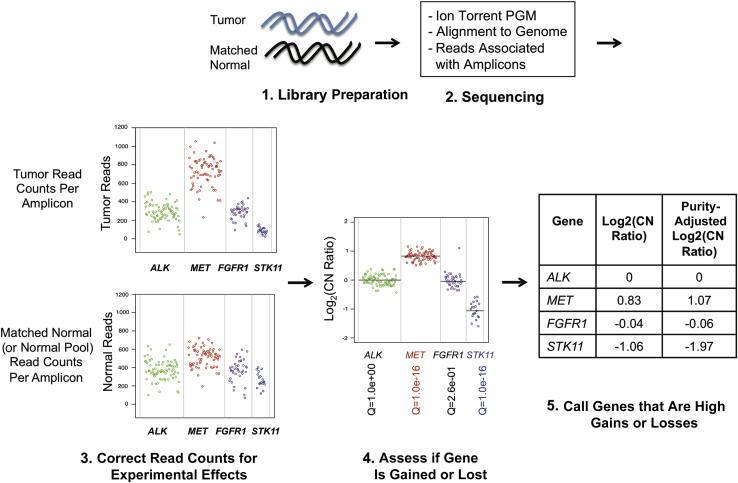
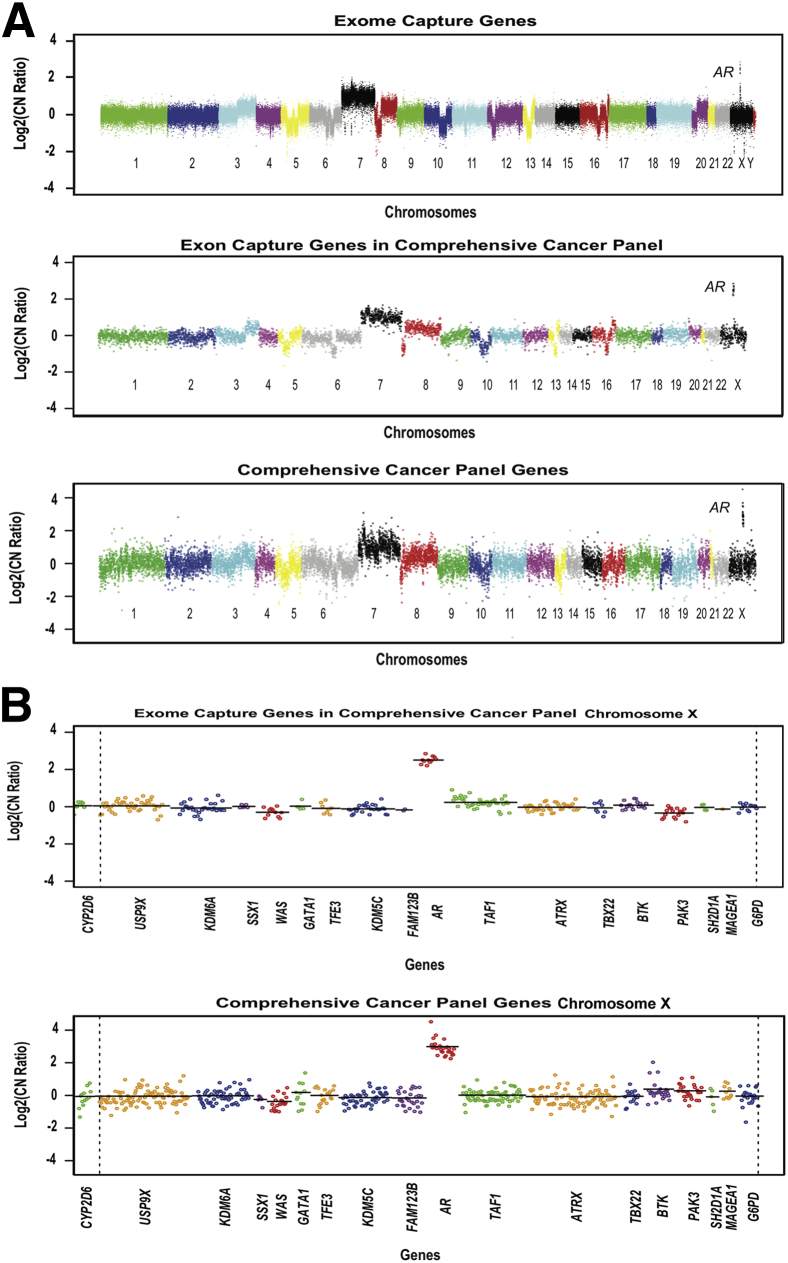
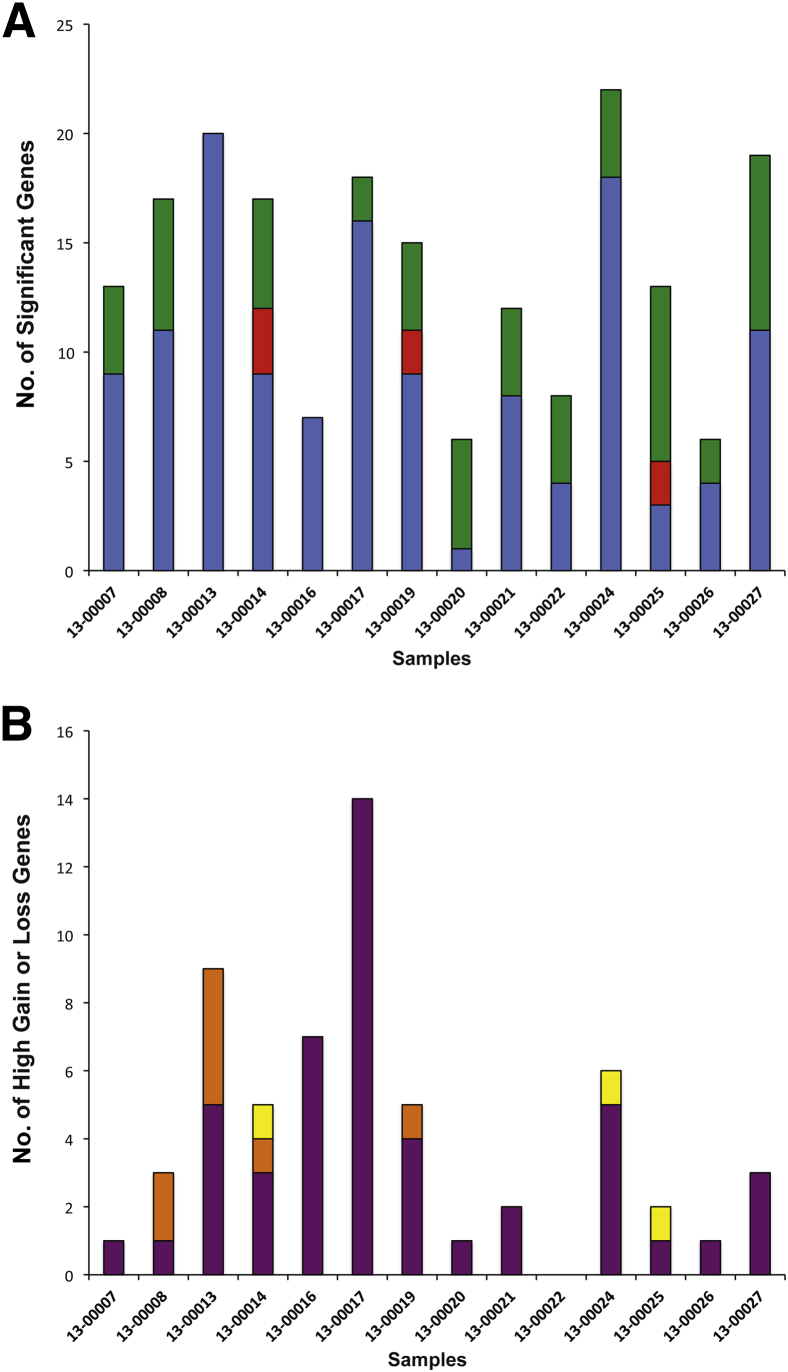
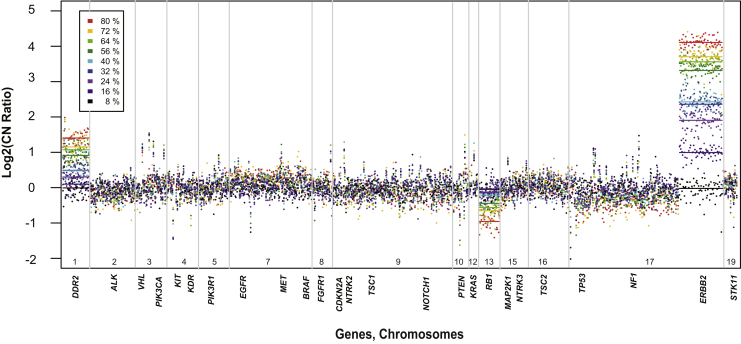
References
-
- Kerick M., Isau M., Timmermann B., Sultmann H., Herwig R., Krobitsch S., Schaefer G., Verdorfer I., Bartsch G., Klocker H., Lehrach H., Schweiger M.R. Targeted high throughput sequencing in clinical cancer settings: formaldehyde fixed-paraffin embedded (FFPE) tumor tissues, input amount and tumor heterogeneity. BMC Med Genomics. 2011;4:68. - PMC - PubMed
-
- Wagle N., Berger M.F., Davis M.J., Blumenstiel B., Defelice M., Pochanard P., Ducar M., Van H.P., Macconaill L.E., Hahn W.C., Meyerson M., Gabriel S.B., Garraway L.A. High-throughput detection of actionable genomic alterations in clinical tumor samples by targeted, massively parallel sequencing. Cancer Discov. 2012;2:82–93. - PMC - PubMed
-
- Beadling C., Neff T.L., Heinrich M.C., Rhodes K., Thornton M., Leamon J., Andersen M., Corless C.L. Combining highly multiplexed PCR with semiconductor-based sequencing for rapid cancer genotyping. J Mol Diagn. 2013;15:171–176. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
