

First human treatment with investigational rhGUS enzyme replacement therapy in an advanced stage MPS VII patient
- PMID: 25468648
- PMCID: PMC4360956
- DOI: 10.1016/j.ymgme.2014.10.017
First human treatment with investigational rhGUS enzyme replacement therapy in an advanced stage MPS VII patient
Abstract
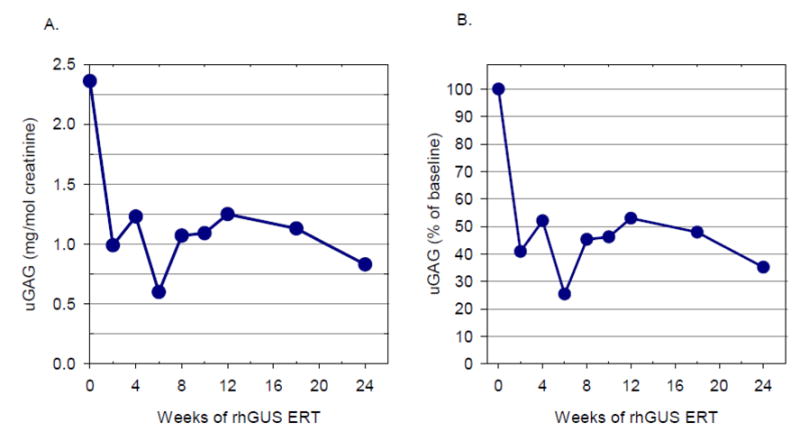
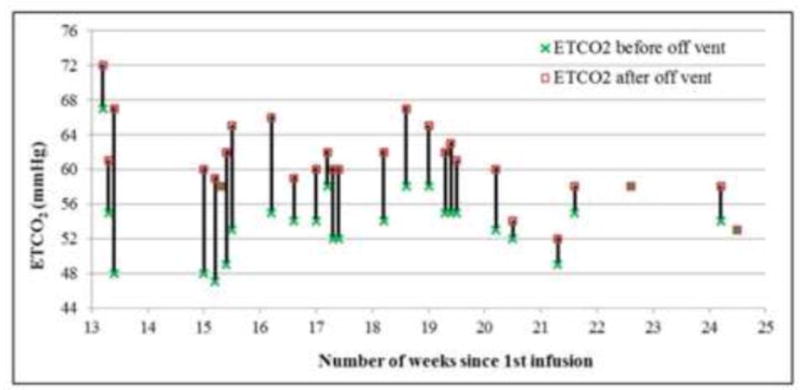
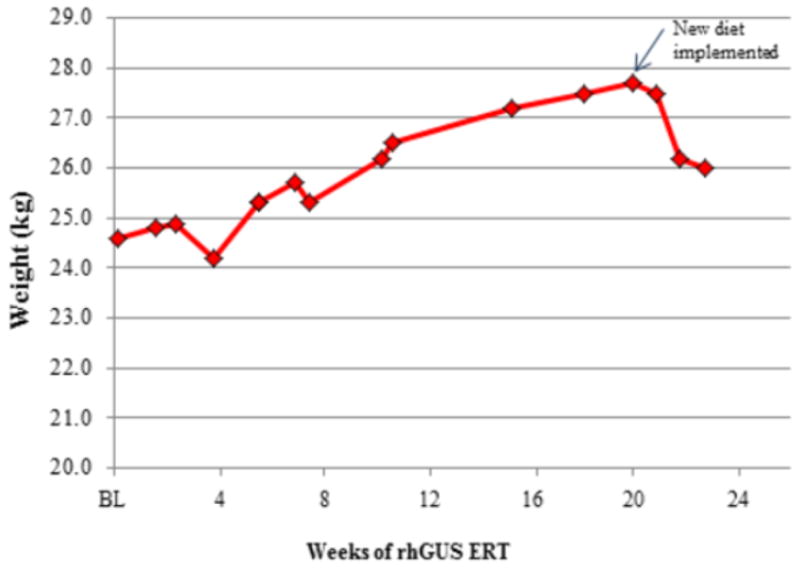
Mucopolysaccharidosis type VII (MPS VII, Sly syndrome) is a very rare lysosomal storage disease caused by a deficiency of the enzyme β-glucuronidase (GUS), which is required for the degradation of three glycosaminoglycans (GAGs): dermatan sulfate, heparan sulfate, and chondroitin sulfate. Progressive accumulation of these GAGs in lysosomes leads to increasing dysfunction in numerous tissues and organs. Enzyme replacement therapy (ERT) has been used successfully for other MPS disorders, but there is no approved treatment for MPS VII. Here we describe the first human treatment with recombinant human GUS (rhGUS), an investigational therapy for MPS VII, in a 12-year old boy with advanced stage MPS VII. Despite a tracheostomy, nocturnal continuous positive airway pressure, and oxygen therapy, significant pulmonary restriction and obstruction led to oxygen dependence and end-tidal carbon dioxide (ETCO2) levels in the 60-80mmHg range, eventually approaching respiratory failure (ETCO2 of 100mmHg) and the need for full-time ventilation. Since no additional medical measures could improve his function, we implemented experimental ERT by infusing rhGUS at 2mg/kg over 4h every 2 weeks for 24 weeks. Safety was evaluated by standard assessments and observance for any infusion associated reactions (IARs). Urinary GAG (uGAG) levels, pulmonary function, oxygen dependence, CO2 levels, cardiac valve function, liver and spleen size, and growth velocity were assessed to evaluate response to therapy. rhGUS infusions were well tolerated. No serious adverse events (SAEs) or IARs were observed. After initiation of rhGUS infusions, the patient's uGAG excretion decreased by more than 50%. Liver and spleen size were reduced within 2 weeks of the first infusion and reached normal size by 24 weeks. Pulmonary function appeared to improve during the course of treatment based on reduced changes in ETCO2 after off-ventilator challenges and a reduced oxygen requirement. The patient regained the ability to eat orally, gained weight, and his energy and activity levels increased. Over 24 weeks, treatment with every-other-week infusions of rhGUS was well tolerated with no SAEs, IARs, or hypersensitivity reactions and was associated with measurable improvement in objective clinical measures and quality of life.
Keywords: Enzyme replacement therapy; Hepatosplenomegaly; Mucopolysaccharidosis type VII; Physician global impression of change; Pulmonary function; Sly syndrome.
Copyright © 2014 Elsevier Inc. All rights reserved.
Conflict of interest statement
Ms Bullaro reports no conflicts of interest.
Figures
References
-
- Neufeld E, Muenzer J. The mucopolysaccharidoses. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The metabolic and molecular bases of inhertited disease. McGraw-Hill, New York: 2001.
-
- Sly WS, Quinton BA, McAlister WH, Rimoin DL. Beta glucuronidase deficiency: Report of clinical, radiologic, and biochemical features of a new mucopolysaccharidosis. J Pediatr. 1973;82:249–257. - PubMed
-
- Vogler C, Levy B, Kyle JW, Sly WS, Williamson J, Whyte MP. Mucopolysaccharidosis vii: Postmortem biochemical and pathological findings in a young adult with beta-glucuronidase deficiency. Mod Pathol. 1994;7:132–137. - PubMed
-
- Harmatz P, Giugliani R, Schwartz I, Guffon N, Teles EL, Miranda MC, Wraith JE, Beck M, Arash L, Scarpa M, Yu ZF, Wittes J, Berger KI, Newman MS, Lowe AM, Kakkis E, Swiedler SJ, M.V.P.S. Group Enzyme replacement therapy for mucopolysaccharidosis vi: A phase 3, randomized, double-blind, placebo-controlled, multinational study of recombinant human n-acetylgalactosamine 4-sulfatase (recombinant human arylsulfatase b or rhasb) and follow-on, open-label extension study. J Pediatr. 2006;148:533–539. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
