

The African Genome Variation Project shapes medical genetics in Africa
- PMID: 25470054
- PMCID: PMC4297536
- DOI: 10.1038/nature13997
The African Genome Variation Project shapes medical genetics in Africa
Abstract
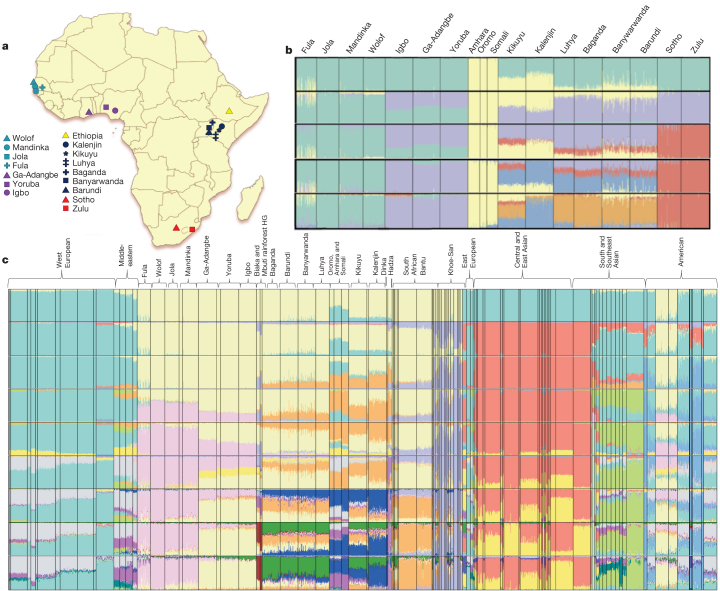
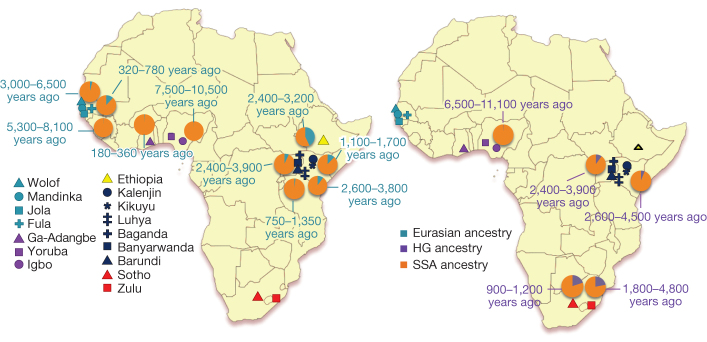
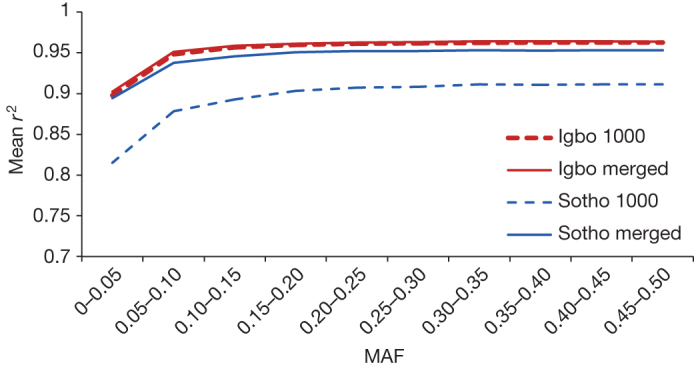
Given the importance of Africa to studies of human origins and disease susceptibility, detailed characterization of African genetic diversity is needed. The African Genome Variation Project provides a resource with which to design, implement and interpret genomic studies in sub-Saharan Africa and worldwide. The African Genome Variation Project represents dense genotypes from 1,481 individuals and whole-genome sequences from 320 individuals across sub-Saharan Africa. Using this resource, we find novel evidence of complex, regionally distinct hunter-gatherer and Eurasian admixture across sub-Saharan Africa. We identify new loci under selection, including loci related to malaria susceptibility and hypertension. We show that modern imputation panels (sets of reference genotypes from which unobserved or missing genotypes in study sets can be inferred) can identify association signals at highly differentiated loci across populations in sub-Saharan Africa. Using whole-genome sequencing, we demonstrate further improvements in imputation accuracy, strengthening the case for large-scale sequencing efforts of diverse African haplotypes. Finally, we present an efficient genotype array design capturing common genetic variation in Africa.
Conflict of interest statement
The authors declare no competing financial interests.
Figures










Comment in
-
Genomics: African dawn.Nature. 2015 Jan 15;517(7534):276-7. doi: 10.1038/nature14077. Epub 2014 Dec 3. Nature. 2015. PMID: 25470066 No abstract available.
-
Population genetics: the African Genome Variation Project.Nat Rev Genet. 2015 Feb;16(2):68-9. doi: 10.1038/nrg3886. Epub 2014 Dec 16. Nat Rev Genet. 2015. PMID: 25511430 No abstract available.
References
-
- The International HapMap Consortium. The International HapMap Project. Nature426, 789–796 (2003) - PubMed
Publication types
MeSH terms
Grants and funding
- MR/K013491/1/MRC_/Medical Research Council/United Kingdom
- Z01 HG200362/ImNIH/Intramural NIH HHS/United States
- P20 MD006899/MD/NIMHD NIH HHS/United States
- 090770/WT_/Wellcome Trust/United Kingdom
- G0901213-92157/MRC_/Medical Research Council/United Kingdom
- G0801566/MRC_/Medical Research Council/United Kingdom
- MC_UP_A900_1118/MRC_/Medical Research Council/United Kingdom
- G1001333/MRC_/Medical Research Council/United Kingdom
- ZIA HG200362/ImNIH/Intramural NIH HHS/United States
- G0600718/MRC_/Medical Research Council/United Kingdom
- 100715/WT_/Wellcome Trust/United Kingdom
- G0901213/MRC_/Medical Research Council/United Kingdom
- 100891/WT_/Wellcome Trust/United Kingdom
- WT077383/Z/05/Z/WT_/Wellcome Trust/United Kingdom
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
