

Effectiveness of exome and genome sequencing guided by acuity of illness for diagnosis of neurodevelopmental disorders
- PMID: 25473036
- PMCID: PMC4286868
- DOI: 10.1126/scitranslmed.3010076
Effectiveness of exome and genome sequencing guided by acuity of illness for diagnosis of neurodevelopmental disorders
Abstract
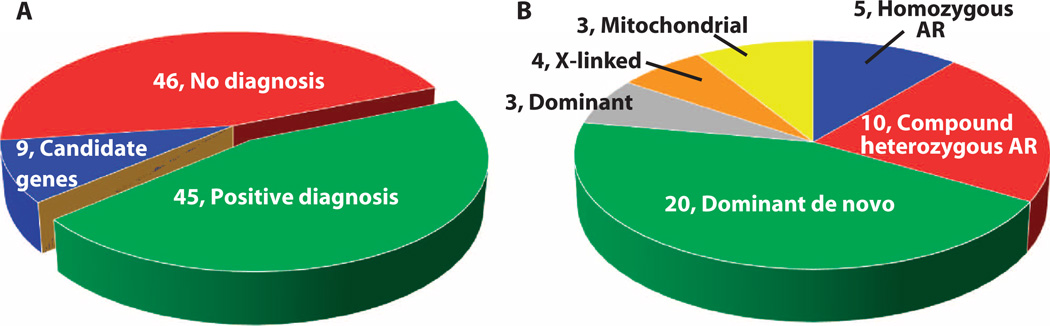
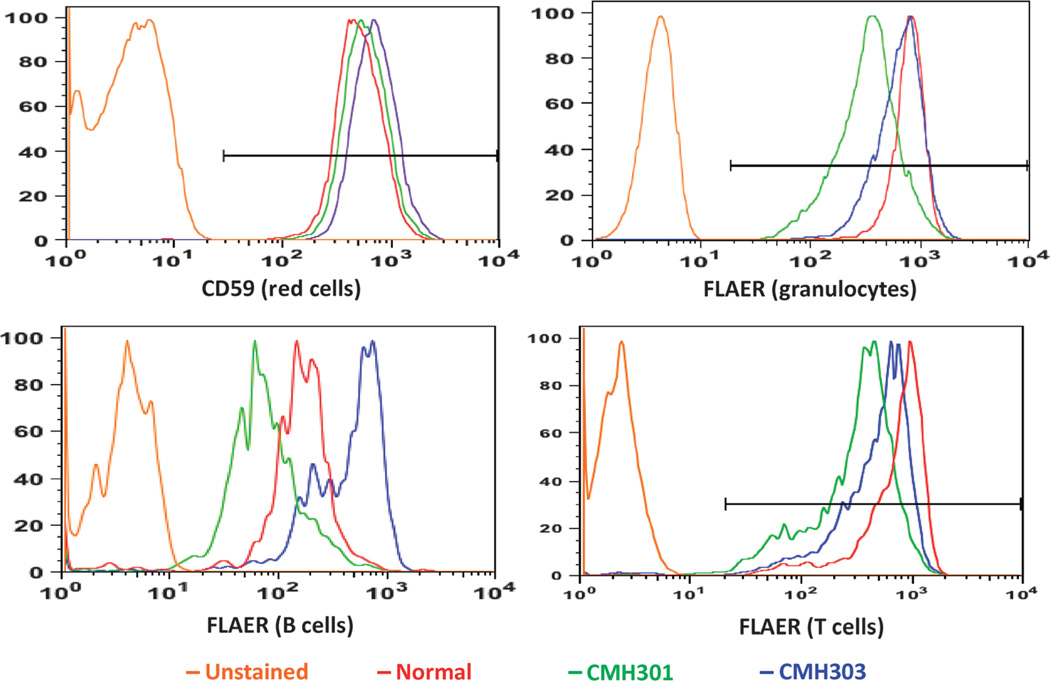
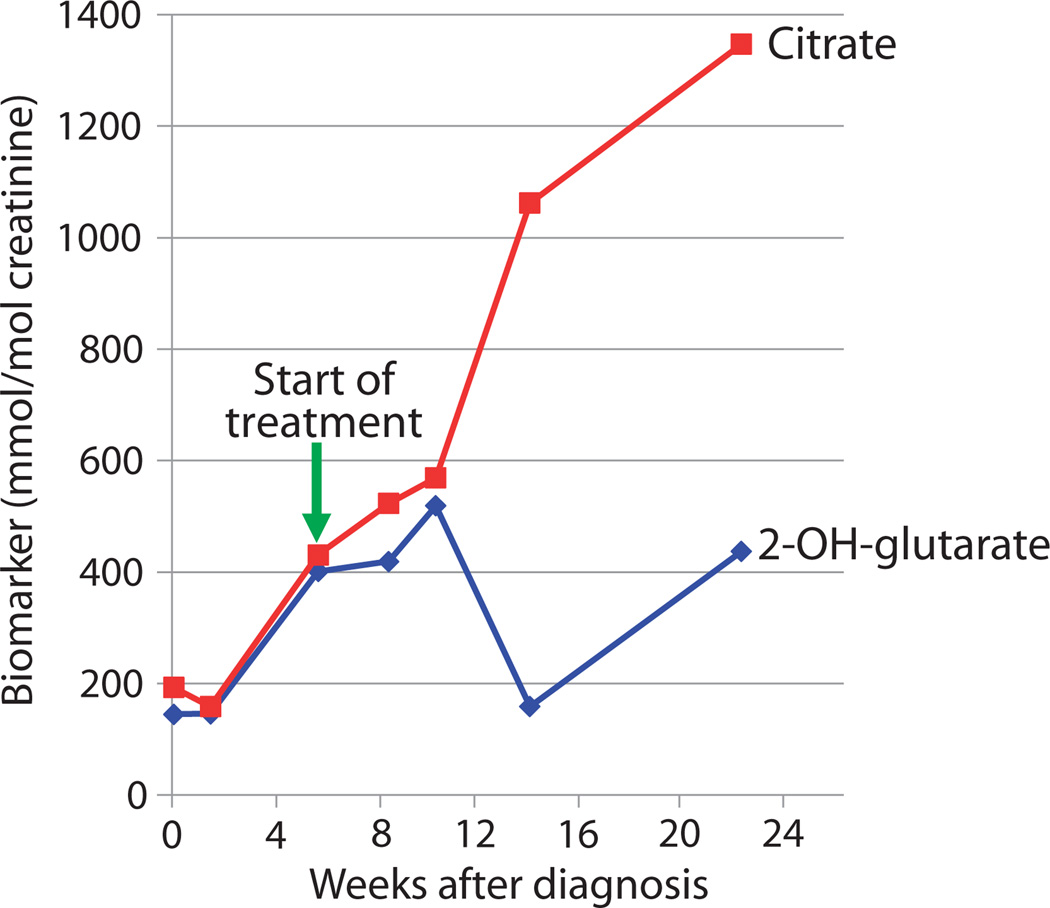
Neurodevelopmental disorders (NDDs) affect more than 3% of children and are attributable to single-gene mutations at more than 1000 loci. Traditional methods yield molecular diagnoses in less than one-half of children with NDD. Whole-genome sequencing (WGS) and whole-exome sequencing (WES) can enable diagnosis of NDD, but their clinical and cost-effectiveness are unknown. One hundred families with 119 children affected by NDD received diagnostic WGS and/or WES of parent-child trios, wherein the sequencing approach was guided by acuity of illness. Forty-five percent received molecular diagnoses. An accelerated sequencing modality, rapid WGS, yielded diagnoses in 73% of families with acutely ill children (11 of 15). Forty percent of families with children with nonacute NDD, followed in ambulatory care clinics (34 of 85), received diagnoses: 33 by WES and 1 by staged WES then WGS. The cost of prior negative tests in the nonacute patients was $19,100 per family, suggesting sequencing to be cost-effective at up to $7640 per family. A change in clinical care or impression of the pathophysiology was reported in 49% of newly diagnosed families. If WES or WGS had been performed at symptom onset, genomic diagnoses may have been made 77 months earlier than occurred in this study. It is suggested that initial diagnostic evaluation of children with NDD should include trio WGS or WES, with extension of accelerated sequencing modalities to high-acuity patients.
Copyright © 2014, American Association for the Advancement of Science.
Figures
Comment in
-
Benefits of genomic sequencing evident in pediatric diagnoses: recent study finds testing method less costly, more effective than other medical, genetic tests.Am J Med Genet A. 2015 Mar;167A(3):vii-viii. doi: 10.1002/ajmg.a.37019. Am J Med Genet A. 2015. PMID: 25691429 No abstract available.
References
-
- Shashi V, McConkie-Rosell A, Rosell B, Schoch K, Vellore K, McDonald M, Jiang YH, Xie P, Need A, Goldstein DB. The utility of the traditional medical genetics diagnostic evaluation in the context of next-generation sequencing for undiagnosed genetic disorders. Genet. Med. 2014;16:176–182. - PubMed
-
- Miller DT, Adam MP, Aradhya S, Biesecker LG, Brothman AR, Carter NP, Church DM, Crolla JA, Eichler EE, Epstein CJ, Faucett WA, Feuk L, Friedman JM, Hamosh A, Jackson L, Kaminsky EB, Kok K, Krantz ID, Kuhn RM, Lee C, Ostell JM, Rosenberg C, Scherer SW, Spinner NB, Stavropoulos DJ, Tepperberg JH, Thorland EC, Vermeesch JR, Waggoner DJ, Watson MS, Martin CL, Ledbetter DH. Consensus statement: Chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies. Am. J. Hum. Genet. 2010;86:749–764. - PMC - PubMed
-
- Battaglia A, Doccini V, Bernardini L, Novelli A, Loddo S, Capalbo A, Filippi T, Carey JC. Confirmation of chromosomal microarray as a first-tier clinical diagnostic test for individuals with developmental delay, intellectual disability, autism spectrum disorders and dysmorphic features. Eur. J. Paediatr. Neurol. 2013;17:589–599. - PubMed
-
- Bartnik M, Nowakowska B, Derwińska K, Wiśniowiecka-Kowalnik B, Kędzior M, Bernaciak J, Ziemkiewicz K, Gambin T, Sykulski M, Bezniakow N, Korniszewski L, Kutkowska-Kaźmierczak A, Klapecki J, Szczałuba K, Shaw CA, Mazurczak T, Gambin A, Obersztyn E, Bocian E, Stankiewicz P. Application of array comparative genomic hybridization in 256 patients with developmental delay or intellectual disability. J. Appl. Genet. 2014;55:125–144. - PMC - PubMed
-
- Kashevarova AA, Nazarenko LP, Skryabin NA, Salyukova OA, Chechetkina NN, Tolmacheva EN, Sazhenova EA, Magini P, Graziano C, Romeo G, Kučinskas V, Lebedev IN. Array CGH analysis of a cohort of Russian patients with intellectual disability. Gene. 2014;536:145–150. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
